Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Chiara Villa.
Facioscapulohumeral muscular dystrophy (FSHD) represents the third most common form of muscular dystrophy and is characterized by muscle weakness and atrophy. FSHD is caused by the altered expression of the transcription factor double homeobox 4 (DUX4), which is involved in several significantly altered pathways required for myogenesis and muscle regeneration.
- facioscapulohumeral muscular dystrophy
- DUX4
- muscle differentiation
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common form of muscular dystrophy after myotomic and Duchenne muscular dystrophy with an estimated incidence of 1:15,000 to 1:20,000 [1]. FSHD affects the facial (facio) and shoulder girdle (scapulo-humeral) muscles, hence the name facioscapulohumeral dystrophy (Figure 1). The disease usually progresses from the upper to lower extremities, with the subsequent involvement of the anterior distal muscles of the leg. Muscle malfunction can impair high-frequency hearing and retinal telangiectasias [2]. Both sexes of all ages can be affected by FSHD [3[3][4],4], although men may develop an earlier onset than women in the case of mosaicism [5,6][5][6]. The symptoms typically appear during adolescence, but the first onset and severity might vary greatly, and in the most severe cases, the symptoms may appear in infancy [7]. Currently, there are no treatments for FSHD, and available therapeutic options include improvements in daily functioning, surveillance for extramuscular complications, and minimizing discomfort and tiredness [8].
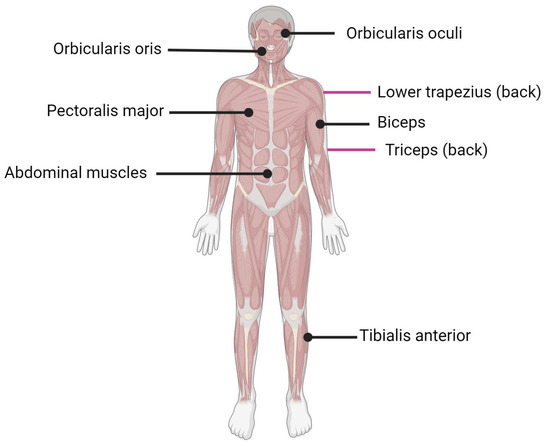
Figure 1. Muscles affected by FSHD. Black lines indicate muscle located in front of the body; purple lines indicate muscle located on the back of the body.
After intensive research, there is a consensus that FSHD is caused by the aberrant expression of the full-length isoform of double homeobox transcription factor (DUX4), particularly in skeletal muscle nuclei [9]. DUX4 is normally expressed in the early stages of development in stem cells and germ lines, especially in the testis, while it is repressed via a repetition-mediated epigenetic silencing (methylation) mechanism [10] during cell differentiation [11] and in most adult somatic tissues, including muscle [12[12][13],13], except for the thymus [14] and keratinocytes [15]. However, the precise mechanism by which this gene induces dystrophic changes, as well as the changes themselves induced at the cellular level, are still controversial and under investigation [12,15][12][15]. Tassin et al., have proposed a dynamic model for DUX4 protein expression in FSHD myotubes. In this model, the DUX4 transcription factor, initially expressed in few nuclei, diffuses in many nuclei of the myotube and thus activates a transcriptional deregulation cascade in each nucleus to which it has diffused. The presence of a dystrophic phenotype causes compromised muscle tissue. The researchers hypothesized that even low levels of DUX4 could result in the formation of amorphous muscle cells [16]. The relatively higher presence of DUX4 in myotubes as opposed to proliferating myoblasts may imply that DUX4 transcription is induced during differentiation.
2. Myogenesis and Muscle Regeneration
Skeletal muscle, one of the three major muscle types, is a contractile tissue responsible for movement, maintaining posture, supporting soft tissues, and maintaining temperature. Tendons are bundles of collagen fibers that connect skeletal muscles to bones, skin, and other muscles. Skeletal muscle is composed of multinuclear cells called myofibers [17], which are formed by the fusion of myoblasts during development [18]. When a muscle is injured, it activates a complex response that leads to tissue regeneration [19,20,21][19][20][21]. Skeletal muscle regeneration is primarily mediated by satellite cells (SCs) that receive signals from the surrounding environment [17,22,23,24][17][22][23][24], which replenish myogenic progenitor cells and differentiate into new myofiber for muscle repair in response to injury [17,25,26][17][25][26]. Muscle regeneration and differentiation are initiated with the modulation of the expression of certain genes and proteins: myogenic regulatory factors (MRFs, summarized in Table 1).Table 1.
Myogenic regulatory factors (MRFs) related to FSHD.
| MRFs | Functions | Reference |
|---|---|---|
| MyoD |
|
[27,28][27][28] |
| Myf5 |
|
[27,28][27][28] |
| MRF4 |
|
[27,28][27][28] |
| Myogenin |
|
[27,28][27][28] |
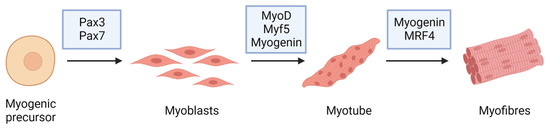
Figure 2.
Myogenic markers and stage-specific expression of the major proteins involved in muscle differentiation.
3. Myoblast Fusion
Myoblast fusion is the process that results in the generation of syncytial muscle cells. It can occur between myoblasts (primary fusion) and myotubes (secondary fusion), and it can also happen during muscle regeneration. Injury is sufficient to activate SCs, which can produce new myoblasts after an asymmetric cellular division, necessary to maintain the SC pool [42]. Mechanistic studies of these components suggest that muscle cells go through at least three consecutive steps before forming a fusion pore [43]. Muscle cell fusion begins when myoblasts exit the cell cycle. Myoblasts will proliferate without differentiating if growth factors (particularly fibroblast growth factors) are present. The second step is cell recognition, which involves aligning the myoblasts into chains. The third step is the cell fusion event itself. Recent studies in a variety of model organisms have uncovered many molecular components required for myoblast fusion. Many steps in this process are facilitated by the actin cytoskeleton, for example. Myoblasts need cytoskeletal shape changes to migrate toward their sites of fusion, and a reorganization of the actin cytoskeleton is required for the following steps: fusion recognition, adhesion, and vesicle transport [44]. Furthermore, although glycolipids and cholesterol are less abundant, they play an important role in regulating membrane polarity and fluidity [45]. Moreover, cholesterol is required for the formation of specialized membrane regions responsible for the regulation of fusion signaling, such as lipid rafts and caveolae [46]. Another fundamental molecule class is represented by the proteins involved in recognition and adhesion. This step necessitates the use of specific integrin family members and cell adhesion molecules (CAMs). The recognition is also mediated by cell membrane glycoproteins, including several cadherins [47]. It plays an important role in mammalian myoblast regeneration, but it has also been found in developing muscle, even if its pathway expression is more visible after the initial fusion steps have been completed. Moreover, M-cadherin (M-cad) is also expressed in SCs and the sarcolemma. Once fusion occurs, M-cad signaling is switched off by M-cad movement into caveolae. M-cad is thus sequestered from the plasma membrane and subsequently transported to the proteasome for degradation [48]. Furthermore, recent studies have identified specific cell signaling pathways whose activation results in the expression of genes required for the fusion process and cytoskeleton rearrangement regulation.4. Genetics of FSHD
On the basis of their underlying epigenetic mechanism, two well-defined subtypes of FSHD exist, namely FSHD1 and FSHD2. Both of them often show an autosomal dominant pattern of inheritance and result in chromatin relaxation and abnormal DUX4 expression in skeletal muscle, leading to progressive muscle weakness and atrophy [49,50][49][50]. FSHD1 represents the most common form, accounting for about 95% of all FSHD cases, and is caused by the partial deletion (shortening or contraction) of the macrosatellite D4Z4 repeat, located in the subtelomeric region of chromosome 4 (4q35) (Figure 3) [16,51,52,53][16][51][52][53]. The D4Z4 macrosatellite, consisting of repeated units of 3.3 kb, is highly polymorphic: in the healthy population, the number of copies varies between 11 and 150, whereas in affected individuals, the number of copies ranges between 1 and 10. This contraction results in a partial loss of D4Z4 DNA methylation, which ultimately leads to DUX4 transcription in skeletal muscle [54]. The severity of the disease increases as the number of repetitions decreases [55,56][55][56]. The decrease in D4Z4 units leads to chromosome relaxation and hypomethylation, allowing DUX4 transcription in muscle cells [53]. In addition to DUX4, additional genes located in the 4q35 region proximal to the D4Z4 repeat array, such as the FSHD region genes 1 and 2 (FRG1, FRG2), adenine nucleotide translocator 1 (ANT1) and FAT atypical cadherin 1 (FAT1), seem to be inappropriately overexpressed in affected muscles [57], but their role in both the onset and severity of disease is still controversial. FRG1 has been considered a candidate gene because of its development of a phenotype similar to that of FSHD in a murine model overexpressing FRG1 [57], and it has been linked to muscle development [10]. Indeed, while FRG1 is subject to extreme variability, it has been found to be upregulated in affected patients. FRG2 is 37kb proximal to D4Z4 and is specifically upregulated in FSHD muscle cells that are differentiating [58]. This gene does not appear to be expressed in some FSHD patients who have an extended deletion at the proximal portion of the macrosatellite, implying that its dysregulation is more likely the result of epigenetic changes than a direct cause of pathology [59,60][59][60]. In support of this, FRG2 overexpression in mouse models did not result in the development of muscular dystrophy [61,62][61][62]. ANT1 encodes a mitochondrial homodimeric protein that is localized asymmetrically on the inner mitochondrial membrane. The dimer forms a membrane channel through which ADP can pass from the matrix to the cytoplasm, being thus essential for cellular oxidative metabolism. ANT1 protein levels appear to be higher in FSHD muscles than in healthy controls or patients with Duchenne muscular dystrophy [57], making muscle cells more susceptible to oxidative stress and apoptosis [63]. While the involvement of FRG1, FRG2, and ANT1 in FSHD pathogenesis is still debated, the role of the FAT1 gene in this disease has been confirmed by independent studies [64,65,66][64][65][66]. FAT1 is a member of the cadherin-like protein family and is involved in the regulation of tissue growth, morphogenesis, and polarity during development [67]. The first association between FAT1 and FSHD was reported in Fat1-deficient mice, which showed muscular and non-muscular phenotypes resembling FSHD symptoms and pathological features [64]. Other authors observed a lower expression of FAT1 in diseased adult muscles than in matched controls, which did not appear to be regulated by DUX4 [65]. They also found that FAT1 is expressed at lower levels in early-stage FSHD-affected muscles compared to later-stage or unaffected muscles in control fetal human biopsies or developing mice embryos [65]. Additional experimental research and case reports have further confirmed FAT1 as a gene involved in disease onset and severity [64,66][64][66]. However, further in-depth studies are needed to clearly understand its role along with the cellular and molecular mechanisms leading to its altered expression in FSHD cells [64,66,68][64][66][68].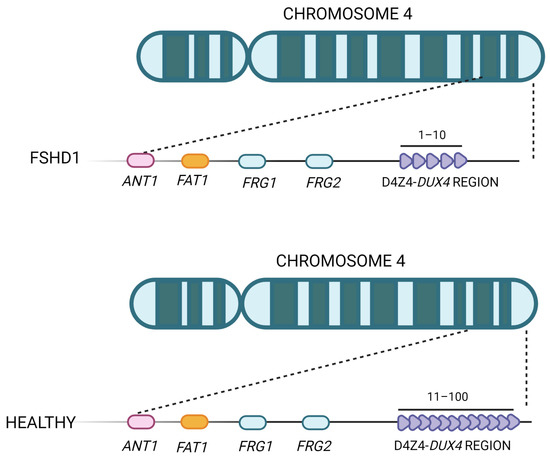
Figure 3. Representation of the FSHD locus. The D4Z4 repeat array is located in the subtelomere of chromosome 4 and can vary between 11 and 100 copies in healthy individuals. In FSHD patients, the structure of D4Z4 adopts a more open configuration and has fewer copies (between 1 and 10).
References
- Hamel, J.; Johnson, N.; Tawil, R.; Martens, W.B.; Dilek, N.; McDermott, M.P.; Heatwole, C. Patient-Reported Symptoms in Facioscapulohumeral Muscular Dystrophy (PRISM-FSHD). Neurology 2019, 93, e1180–e1192.
- Caputo, V.; Megalizzi, D.; Fabrizio, C.; Termine, A.; Colantoni, L.; Caltagirone, C.; Giardina, E.; Cascella, R.; Strafella, C. Update on the Molecular Aspects and Methods Underlying the Complex Architecture of FSHD. Cells 2022, 11, 2687.
- Himeda, C.L.; Jones, P.L. FSHD Therapeutic Strategies: What Will It Take to Get to Clinic? J. Pers. Med. 2022, 12, 865.
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66.
- Zatz, M.; Marie, S.K.; Cerqueira, A.; Vainzof, M.; Pavanello, R.C.; Passos-Bueno, M.R. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am. J. Med. Genet. 1998, 77, 155–161.
- Van der Maarel, S.M.; Deidda, G.; Lemmers, R.J.; van Overveld, P.G.; van der Wielen, M.; Hewitt, J.E.; Sandkuijl, L.; Bakker, B.; van Ommen, G.J.; Padberg, G.W.; et al. De novo facioscapulohumeral muscular dystrophy: Frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am. J. Hum. Genet. 2000, 66, 26–35.
- Chen, T.H.; Wu, Y.Z.; Tseng, Y.H. Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review. Int. J. Mol. Sci. 2020, 21, 7783.
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 85, 357–364.
- Tawil, R.; McDermott, M.P.; Mendell, J.R.; Kissel, J.; Griggs, R.C. Facioscapulohumeral muscular dystrophy (FSHD): Design of natural history study and results of baseline testing. FSH-DY Group. Neurology 1994, 44, 442–446.
- Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. Facioscapulohumeral dystrophy: The path to consensus on pathophysiology. Skelet. Muscle 2014, 4, 12.
- Sacconi, S.; Salviati, L.; Desnuelle, C. Facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta 2015, 1852, 607–614.
- Zernov, N.; Skoblov, M. Genotype-phenotype correlations in FSHD. BMC Med. Genom. 2019, 12, 43.
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181.
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022.
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 2018, 15, 863–871.
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013, 17, 76–89.
- Fukada, S.I. The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J. Biochem. 2018, 163, 353–358.
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462.
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062.
- Barthélémy, F.; Wein, N. Personalized gene and cell therapy for Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2018, 28, 803–824.
- Morgan, J.; Partridge, T. Skeletal muscle in health and disease. Dis. Model. Mech. 2020, 13, dmm042192.
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581.
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646.
- Yamamoto, M.; Legendre, N.P.; Biswas, A.A.; Lawton, A.; Yamamoto, S.; Tajbakhsh, S.; Kardon, G.; Goldhamer, D.J. Loss of MyoD and Myf5 in Skeletal Muscle Stem Cells Results in Altered Myogenic Programming and Failed Regeneration. Stem Cell Rep. 2018, 10, 956–969.
- Relaix, F.; Marcelle, C. Muscle stem cells. Curr. Opin. Cell Biol. 2009, 21, 748–753.
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011, 138, 3657–3666.
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18.
- Shirakawa, T.; Toyono, T.; Inoue, A.; Matsubara, T.; Kawamoto, T.; Kokabu, S. Factors Regulating or Regulated by Myogenic Regulatory Factors in Skeletal Muscle Stem Cells. Cells 2022, 11, 1493.
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342.
- Świerczek, B.; Ciemerych, M.A.; Archacka, K. From pluripotency to myogenesis: A multistep process in the dish. J. Muscle Res. Cell. Motil. 2015, 36, 363–375.
- Hauerslev, S.; Vissing, J.; Krag, T.O. Muscle atrophy reversed by growth factor activation of satellite cells in a mouse muscle atrophy model. PLoS ONE 2014, 9, e100594.
- Koganti, P.; Yao, J.; Cleveland, B.M. Molecular Mechanisms Regulating Muscle Plasticity in Fish. Animals 2020, 11, 61.
- Kaczmarek, A.; Kaczmarek, M.; Ciałowicz, M.; Clemente, F.M.; Wolański, P.; Badicu, G.; Murawska-Ciałowicz, E. The Role of Satellite Cells in Skeletal Muscle Regeneration-The Effect of Exercise and Age. Biology 2021, 10, 1056.
- Sheveleva, O.N.; Payushina, O.V.; Butorina, N.N.; Domaratskaya, E.I. The Myogenic Potential of Mesenchymal Stromal Cells and Their Effect on Skeletal Muscle Regeneration. Biol. Bull. Russ. Acad. Sci. 2020, 47, 455–465.
- Fochi, S.; Giuriato, G.; De Simone, T.; Gomez-Lira, M.; Tamburin, S.; Del Piccolo, L.; Schena, F.; Venturelli, M.; Romanelli, M.G. Regulation of microRNAs in Satellite Cell Renewal, Muscle Function, Sarcopenia and the Role of Exercise. Int. J. Mol. Sci. 2020, 21, 6732.
- Forcina, L.; Cosentino, M.; Musarò, A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells 2020, 9, 1297.
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122.
- Yusuf, F.; Brand-Saberi, B. Myogenesis and muscle regeneration. Histochem. Cell Biol. 2012, 138, 187–199.
- Doyonnas, R.; LaBarge, M.A.; Sacco, A.; Charlton, C.; Blau, H.M. Hematopoietic contribution to skeletal muscle regeneration by myelomonocytic precursors. Proc. Natl. Acad. Sci. USA 2004, 101, 13507–13512.
- Mourkioti, F.; Rosenthal, N. IGF-1, inflammation and stem cells: Interactions during muscle regeneration. Trends Immunol. 2005, 26, 535–542.
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67.
- Romagnoli, C.; Iantomasi, T.; Brandi, M.L. Available In Vitro Models for Human Satellite Cells from Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 13221.
- Lehka, L.; Rędowicz, M.J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92.
- Millay, D.P. Regulation of the myoblast fusion reaction for muscle development, regeneration, and adaptations. Exp. Cell Res. 2022, 415, 113134.
- Deleu, M.; Crowet, J.M.; Nasir, M.N.; Lins, L. Complementary biophysical tools to investigate lipid specificity in the interaction between bioactive molecules and the plasma membrane: A review. Biochim. Biophys. Acta 2014, 1838, 3171–3190.
- Mukai, A.; Kurisaki, T.; Sato, S.B.; Kobayashi, T.; Kondoh, G.; Hashimoto, N. Dynamic clustering and dispersion of lipid rafts contribute to fusion competence of myogenic cells. Exp. Cell Res. 2009, 315, 3052–3063.
- Schwander, M.; Leu, M.; Stumm, M.; Dorchies, O.M.; Ruegg, U.T.; Schittny, J.; Müller, U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev. Cell 2003, 4, 673–685.
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96.
- Van der Maarel, S.M.; Miller, D.G.; Tawil, R.; Filippova, G.N.; Tapscott, S.J. Facioscapulohumeral muscular dystrophy: Consequences of chromatin relaxation. Curr. Opin. Neurol. 2012, 25, 614–620.
- Gaillard, M.C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014, 83, 733–742.
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162.
- Bosnakovski, D.; Lamb, S.; Simsek, T.; Xu, Z.; Belayew, A.; Perlingeiro, R.; Kyba, M. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 2008, 214, 87–96.
- Van der Maarel, S.M.; Tawil, R.; Tapscott, S.J. Facioscapulohumeral muscular dystrophy and DUX4: Breaking the silence. Trends Mol. Med. 2011, 17, 252–258.
- Van Overveld, P.G.; Lemmers, R.J.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317.
- Greco, A.; Goossens, R.; van Engelen, B.; van der Maarel, S.M. Consequences of epigenetic derepression in facioscapulohumeral muscular dystrophy. Clin. Genet. 2020, 97, 799–814.
- Ehrlich, M.; Jackson, K.; Tsumagari, K.; Camaño, P.; Lemmers, R.J. Hybridization analysis of D4Z4 repeat arrays linked to FSHD. Chromosoma 2007, 116, 107–116.
- Gabellini, D.; Green, M.R.; Tupler, R. Inappropriate gene activation in FSHD: A repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 2002, 110, 339–348.
- Rijkers, T.; Deidda, G.; van Koningsbruggen, S.; van Geel, M.; Lemmers, R.J.; van Deutekom, J.C.; Figlewicz, D.; Hewitt, J.E.; Padberg, G.W.; Frants, R.R.; et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J. Med. Genet. 2004, 41, 826–836.
- Tawil, R.; Van Der Maarel, S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15.
- De Greef, J.C.; Frants, R.R.; van der Maarel, S.M. Epigenetic mechanisms of facioscapulohumeral muscular dystrophy. Mutat. Res. 2008, 647, 94–102.
- Lemmers, R.J.; Osborn, M.; Haaf, T.; Rogers, M.; Frants, R.R.; Padberg, G.W.; Cooper, D.N.; van der Maarel, S.M.; Upadhyaya, M. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: Phenotype, size, and detection. Neurology 2003, 61, 178–183.
- Klooster, R.; Straasheijm, K.; Shah, B.; Sowden, J.; Frants, R.; Thornton, C.; Tawil, R.; van der Maarel, S. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur. J. Hum. Genet. 2009, 17, 1615–1624.
- Arbogast, S.; Kotzur, H.; Frank, C.; Compagnone, N.; Sutra, T.; Pillard, F.; Pietri, S.; Hmada, N.; Moussa, D.M.A.; Bride, J.; et al. ANT1 overexpression models: Some similarities with facioscapulohumeral muscular dystrophy. Redox Biol. 2022, 56, 102450.
- Caruso, N.; Herberth, B.; Bartoli, M.; Puppo, F.; Dumonceaux, J.; Zimmermann, A.; Denadai, S.; Lebossé, M.; Roche, S.; Geng, L.; et al. Deregulation of the protocadherin gene FAT1 alters muscle shapes: Implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet. 2013, 9, e1003550.
- Mariot, V.; Roche, S.; Hourdé, C.; Portilho, D.; Sacconi, S.; Puppo, F.; Duguez, S.; Rameau, P.; Caruso, N.; Delezoide, A.L.; et al. Correlation between low FAT1 expression and early affected muscle in facioscapulohumeral muscular dystrophy. Ann. Neurol. 2015, 78, 387–400.
- Park, H.J.; Lee, W.; Kim, S.H.; Lee, J.H.; Shin, H.Y.; Kim, S.M.; Park, K.D.; Choi, Y.C. FAT1 Gene Alteration in Facioscapulohumeral Muscular Dystrophy Type 1. Yonsei Med. J. 2018, 59, 337–340.
- Saburi, S.; Hester, I.; Goodrich, L.; McNeill, H. Functional interactions between Fat family cadherins in tissue morphogenesis and planar polarity. Development 2012, 139, 1806–1820.
- Jia, F.F.; Drew, A.P.; Nicholson, G.A.; Corbett, A.; Kumar, K.R. Facioscapulohumeral muscular dystrophy type 2: An update on the clinical, genetic, and molecular findings. Neuromuscul. Disord. 2021, 31, 1101–1112.
- Gilbert, J.R.; Stajich, J.M.; Wall, S.; Carter, S.C.; Qiu, H.; Vance, J.M.; Stewart, C.S.; Speer, M.C.; Pufky, J.; Yamaoka, L.H.; et al. Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD). Am. J. Hum. Genet. 1993, 53, 401–408.
- Steel, D.; Main, M.; Manzur, A.; Muntoni, F.; Munot, P. Clinical features of facioscapulohumeral muscular dystrophy 1 in childhood. Dev. Med. Child. Neurol. 2019, 61, 964–971.
- Sacconi, S.; Lemmers, R.J.; Balog, J.; van der Vliet, P.J.; Lahaut, P.; van Nieuwenhuizen, M.P.; Straasheijm, K.R.; Debipersad, R.D.; Vos-Versteeg, M.; Salviati, L.; et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 2013, 93, 744–751.
- Larsen, M.; Rost, S.; El Hajj, N.; Ferbert, A.; Deschauer, M.; Walter, M.C.; Schoser, B.; Tacik, P.; Kress, W.; Müller, C.R. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 2015, 23, 808–816.
- Sacconi, S.; Briand-Suleau, A.; Gros, M.; Baudoin, C.; Lemmers, R.; Rondeau, S.; Lagha, N.; Nigumann, P.; Cambieri, C.; Puma, A.; et al. FSHD1 and FSHD2 form a disease continuum. Neurology 2019, 92, e2273–e2285.
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029.
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417.
- Lemmers, R.; van der Vliet, P.J.; Blatnik, A.; Balog, J.; Zidar, J.; Henderson, D.; Goselink, R.; Tapscott, S.J.; Voermans, N.C.; Tawil, R.; et al. Chromosome 10q-linked FSHD identifies DUX4 as principal disease gene. J. Med. Genet. 2022, 59, 180–188.
- Van Deutekom, J.C.; Bakker, E.; Lemmers, R.J.; van der Wielen, M.J.; Bik, E.; Hofker, M.H.; Padberg, G.W.; Frants, R.R. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: Implications for genetic counselling and etiology of FSHD1. Hum. Mol. Genet. 1996, 5, 1997–2003.
- Nguyen, K.; Broucqsault, N.; Chaix, C.; Roche, S.; Robin, J.D.; Vovan, C.; Gerard, L.; Mégarbané, A.; Urtizberea, J.A.; Bellance, R.; et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. J. Med. Genet. 2019, 56, 590–601.
- Goossens, R.; van den Boogaard, M.L.; Lemmers, R.; Balog, J.; van der Vliet, P.J.; Willemsen, I.M.; Schouten, J.; Maggio, I.; van der Stoep, N.; Hoeben, R.C.; et al. Intronic SMCHD1 variants in FSHD: Testing the potential for CRISPR-Cas9 genome editing. J. Med. Genet. 2019, 56, 828–837.
- Lemmers, R.; van der Vliet, P.J.; Granado, D.S.L.; van der Stoep, N.; Buermans, H.; van Schendel, R.; Schimmel, J.; de Visser, M.; van Coster, R.; Jeanpierre, M.; et al. High-resolution breakpoint junction mapping of proximally extended D4Z4 deletions in FSHD1 reveals evidence for a founder effect. Hum. Mol. Genet. 2022, 31, 748–760.
- Lemmers, R.; van der Vliet, P.J.; Vreijling, J.P.; Henderson, D.; van der Stoep, N.; Voermans, N.; van Engelen, B.; Baas, F.; Sacconi, S.; Tawil, R.; et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum. Mol. Genet. 2018, 27, 3488–3497.
- Van den Boogaard, M.L.; Lemmers, R.J.; Camaño, P.; van der Vliet, P.J.; Voermans, N.; van Engelen, B.G.; Lopez de Munain, A.; Tapscott, S.J.; van der Stoep, N.; Tawil, R.; et al. Double SMCHD1 variants in FSHD2: The synergistic effect of two SMCHD1 variants on D4Z4 hypomethylation and disease penetrance in FSHD2. Eur. J. Hum. Genet. 2016, 24, 78–85.
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653.
- Daxinger, L.; Tapscott, S.J.; van der Maarel, S.M. Genetic and epigenetic contributors to FSHD. Curr. Opin. Genet. Dev. 2015, 33, 56–61.
More