Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Kathleen I. Moineau-Chane Ching.
The research for efficient organic materials organized in bulk heterojunction (BHJ) thin films for organic photovoltaics (OPVs) has shown a significant breakthrough. Desired structural organization can be attained through various strategies. Conjugated polymer-based bulk heterojunction solar cells have exhibited high power conversion efficiencies (PCEs). The role of side chain engineering in solution-processable conjugated polymers is taking a growing role in these record performances.
- alkyl side chains
- organic materials
- organic photovoltaics
- π-conjugated materials
- blend morphology
1. Introduction
Since the first use of conjugated polymers in organic electronics [1], research on their design has led to impressive progress of OPV performances. Numerous hundreds of publications have reported the synthesis, photophysical properties, and use of conjugated polymers in OPVs. Several reviews have gathered these papers [2][3][4], emphasizing the polymerization strategy [5], the role of specific building blocks [6][7][8][9], the improvement of the active layer or cell technology [10][11][12], the achievement of transparent BHJ photoactive layers and devices [2][13], or their morphological design strategies for improving charge carrier mobilities [14]. Recently, conjugated polymer-based bulk heterojunction solar cells have exhibited high power conversion efficiencies (PCEs) up to 18.22% in 2020 [15] and up to 19.3% in a single-junction cell in 2022 [16]. The role of side chain engineering in solution-processable conjugated polymers is taking a growing role in these record performances. The side chains may be of various chemical types, namely, alkyl, hybrid, oligoether, fluoroalkyl, and latently reactive side chains, as detailed in the first review devoted to side chain engineering for polymer-based solar cells [17]. The following sections will focus on the three first classes. The structures of the polymers that are subjects of this discussion are represented in Figure 1. The n-type organic semiconductors evoked in this part are represented in Figure 2.
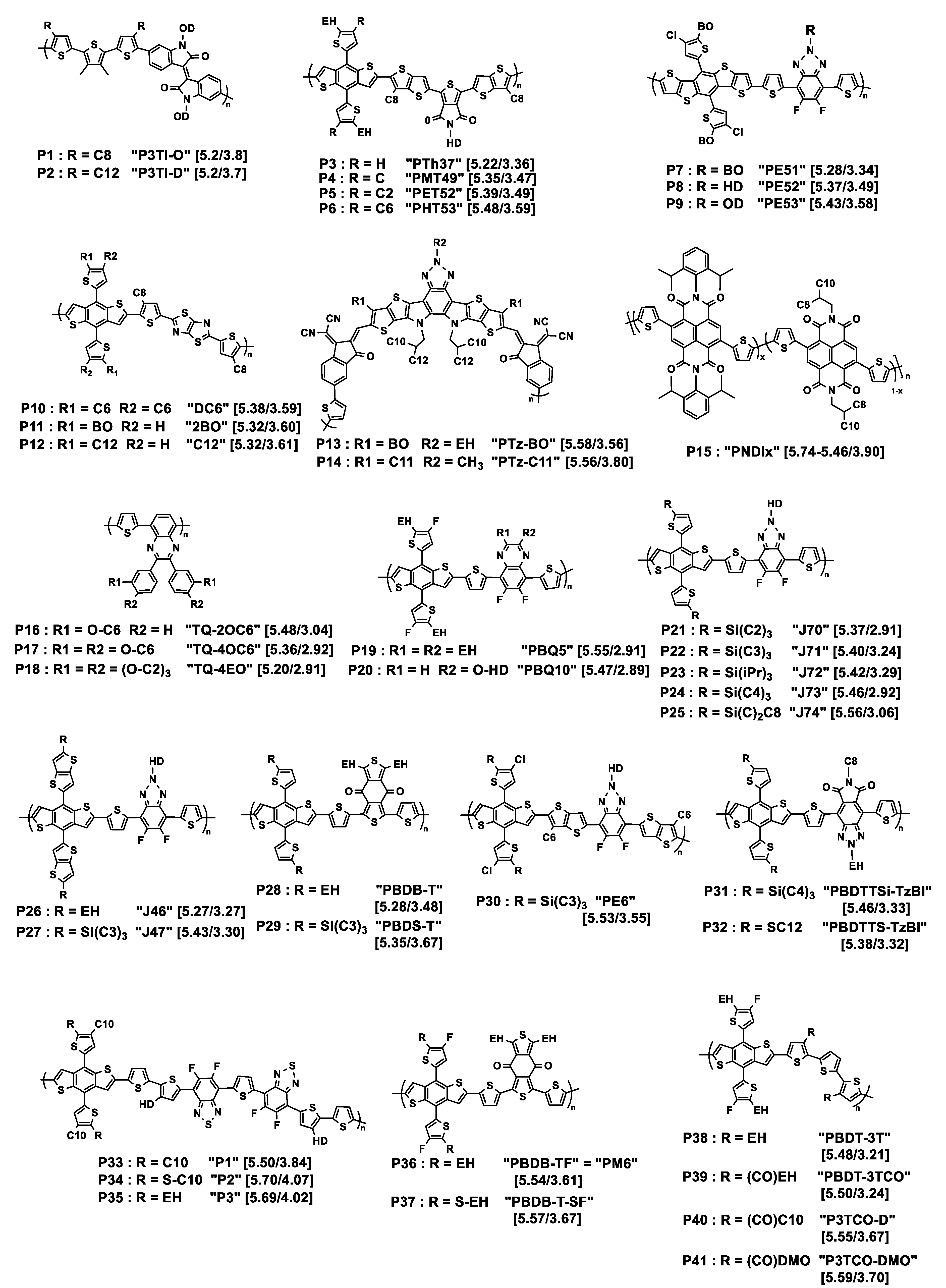
Figure 1. Chemical structures of the donor polymers discussed herein. Px is the abbreviated name of the polymer whose original name is also given in quotation marks. HOMO/LUMO energy levels (eV) are given in square brackets. EH = 2-ethylhexyl; BO = 2-butyloctyl; HD = 2-hexyldecyl; OD = 2-octyldodecyl; DMO = 3,7-dimethyloctyl; iPr = isopropyl. Cx = n-alkyl linear chain with x methylene units.
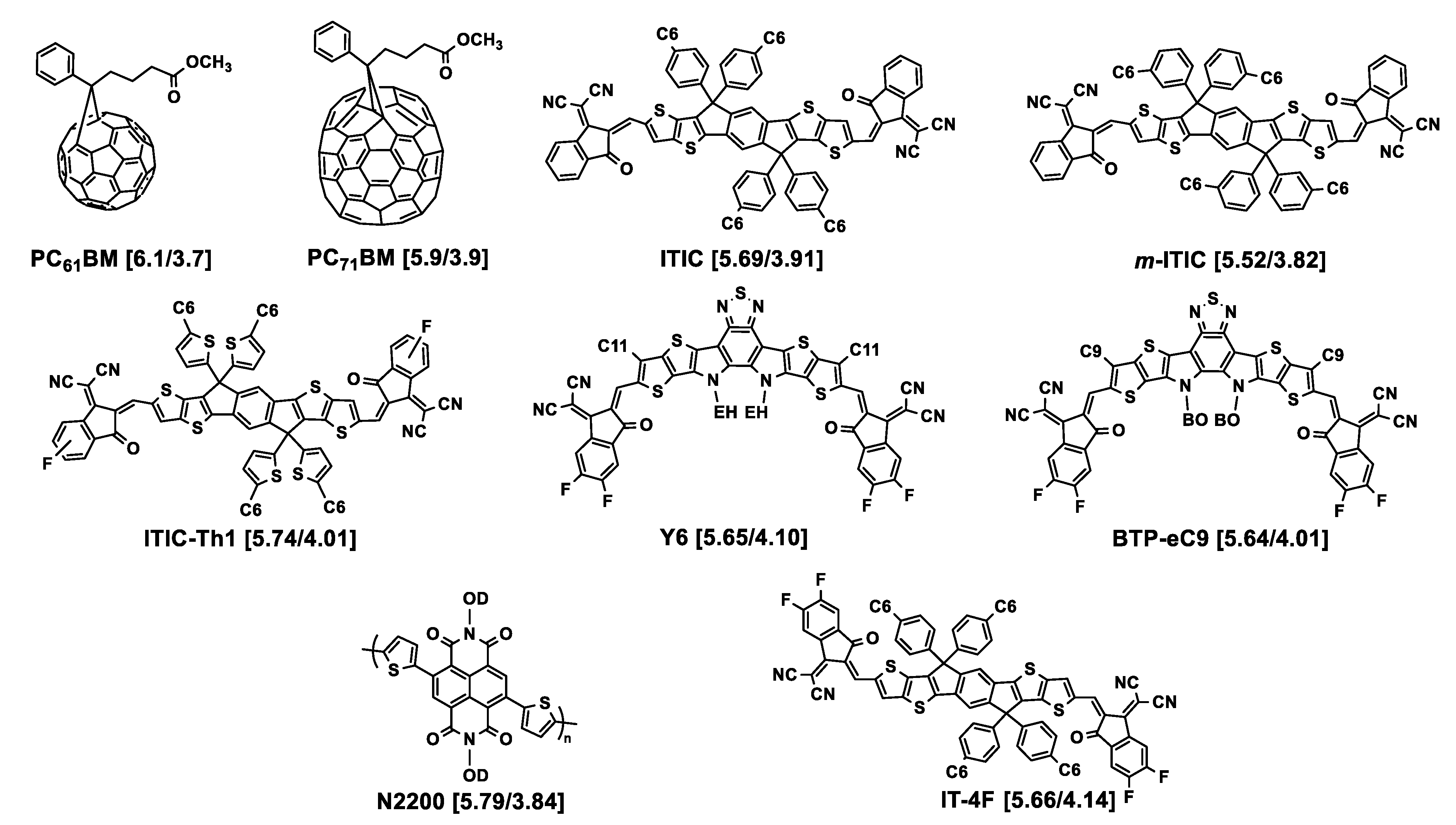
Figure 2. Chemical structures of the acceptors used in association with the polymers described in this section. HOMO/LUMO energy levels (eV) are given in square brackets. EH = 2-ethylhexyl; BO = 2-butyloctyl; OD = 2-octyldodecyl. Cx = n-alkyl linear chain with x methylene units.
It has been very recently pointed out that in polymer systems, one of the main challenges is to obtain efficient and stable BHJ morphological states as soon as the thin film is elaborated, both at the laboratory scale in using spin coating techniques and on an industrial scale where printing methods are preferred [18]. In the first case, the deposition of an already prepared blend of donor and acceptor materials affords a BHJ in which an ideal compromise between D/A miscibility and phase separation is required for nanofibrillar structures [16][19]. In the second case, a two-step sequential deposition method proved to favor the formation of fibril structure of donor polymer in the first layer in which NFA molecules can diffuse into the fibril mesh to form a bicontinuous network morphology [18]. A low miscibility of both materials is detrimental to morphology stability and may be responsible for spontaneous spinodal D/A demixing, as a major issue in BHJ morphology degradation [20]. For investigating blends’ morphology, spectroscopic means such as UV–visible (UV–Vis) spectroscopy are commonly used on solutions and thin films, as well as high-resolution techniques: while the polymer packing behavior may be investigated by grazing-incidence wide-angle X-ray scattering (GIWAXS), that has emerged as a powerful tool in the past two decades for the study of nanostructuration in thin films, atomic force microscopy (AFM) and transmission electron microscopy (TEM) are also used for analyzing the impact of morphology on JSC and FF.
2. Length of n-Alkyl Chains
-
Tegene and collaborators have prepared two polymers based on terthiophene-isoindigo bricks [21]. While the indigo unit is functionalized on the N atoms by bulky 2-octyldodecyl branched chains, the authors have grafted n-alkyl chains of two different lengths onto the terthiophene unit, a C8 and a C12 chain (see P1, P2 in Figure 1), in order to probe the effect of their length on the early photoinduced charge-transfer process. Whereas the chain length had a negligible effect on absorption and emission (steady-state measurements) in solution and thin films, femtosecond transient absorption (fs-TA) spectroscopy revealed that the intramolecular charge-transfer process was three times faster in going from a C12 to a C8 chain. Moreover, exciton–exciton annihilation (EEA) measurements together with estimation of exciton density demonstrated that the exciton diffusion was boosted when the alkyl side chain was shorter. The authors suggested that these results were related to better intra- and interchain interaction in the polymer with shorter alkyl chains. The model case with no alkyl chain on the terthiophene moiety was not reported.
-
Playing with shorter chains, Zhang and collaborators explored the effect of length of linear alkyl chains from C1 to C6 in the 4-position of the pendant thiophene moiety of the 4,8-di(thien-2-yl)benzo [1,2-b:4,5-b′]dithiophene (DTBDT) unit, including the case of no chain being grafted, of polymers resulting from the polycondensation of the benzo [1,2-b;4,5-b′]dithiophene (BDT)-based monomers with 5-(2-ethylhexyl)-1,3-bis(3-octylthieno [3,2-b]thiophen-5-yl)-5H-thieno [3,4-c]pyrrole-4,6-dione (tt-TPD) monomers [22]. The structures of the corresponding polymers P3 to P6 are shown in Figure 1. While they named the polymer without grafted alkyl chain PTh37, PMT49, PET52, and PHT53 were those with methyl, ethyl, and n-hexyl chains on the 4-position of the pendant thiophene moiety, respectively. All the polymers are substituted in the 5-position by an ethylhexyl branched chain, thus situated in the vicinity of the linear chain. It was observed by UV–Vis spectroscopy that the optical bandgaps were 1.86, 1.88, 1.90, and 1.89 eV for PTh37, PMT49, PET52, and PHT53, respectively, demonstrating low impact of the alkyl chains. However, comparison of the thin film spectra revealed a stronger vibrational character of the 500 to 640 nm absorption band with increasing the length of the alkyl chain, suggesting enhanced intermolecular interactions with alkyl chain growth. The photovoltaic parameters of devices prepared with ITIC as the acceptor demonstrated that the methylated polymer PMT49 afforded the best performances, delivering a PCE of 12.1%, against 10.9%, 9.94%, and 8.05% for devices based on PTh37, PET52, and PHT53, respectively. The JSC values evolved in the same way, from 18.52 mA cm−2 for PMT49:ITIC to 17.63 mA cm−2, 15.74 cm−2, and 13.69 cm−2 for PTh37:ITIC, PET52:ITIC and PHT53:ITIC, respectively. Studies performed on thin pristine films and their blends with ITIC confirmed the advantage of PMT49 over the other polymers with higher charge mobilities in blend films and more optimized morphology with smaller domain size (13.09 nm vs. 16.68 nm, 14.67 nm, and 17.38 nm for PTh37:ITIC, PET52:ITIC, and PHT53:ITIC blends, respectively). In that case, it is noticeable that, even with an ethylhexyl branched chain in the 5-position, methyl short chains can finely tune the device performances by exerting a significant influence on the morphology of the resulting polymer and its blends with the acceptor material. Here again, using shorter chains was proved to be beneficial.
3. Length and Linear/Branched Nature of Alkyl Chains
As seen before, there is a compromise in polymer systems between strong self-aggregation phenomena resulting from π–π stacking of the conjugated backbone and reduction of aggregation tendency by using bulky side chains. If we push too far to extremes, in the first case large aggregated domains appear in the film which reduces the polymer miscibility with acceptor materials and, in the second case, the propensity to self-aggregate becomes so weak that the crystallinity of the polymer vanishes, these two cases being causes of phase separation between donor and acceptor [23]. BHJ morphology is linked both to the chemical nature of the donor and acceptor conjugated units forming the polymer backbone and to the substitution sites, the length, or the branching position of the solubilizing side chains. It is often observed that branched alkyl chains positively affect the morphology of thin film in comparison to their linear counterparts, due to their double effect of solubilization and aggregation hindering related to their bulkiness. Tuning this last parameter may influence the way of stacking of conjugated polymer and thus the device performances. A convenient means for this is changing the length of the alkyl chains before and/or after the branching point as reported in several papers.
-
Zhou and coworkers prepared three polymers named as PE51, PE52, and PE53 possessing the same polymer backbone but branched alkyl chains of increasing bulkiness from PE51 to PE53, as presented in Figure 1 (polymers P7–P9) [24]. The backbone is constituted by alternating donor DTBDT and acceptor benzotriazole (BTA) moieties, the latter being substituted on the triazole ring by branched alkyl chains, namely 2-butyloctyl, 2-hexyldecyl, and 2-octyldodecyl. UV–Vis absorption spectra show that, besides the classical charge-transfer transition absorption peak, the three polymers exhibit vibrational absorption peaks in solution, which is related to intramolecular aggregation. After mixing them with Y6 as the acceptor, the resulting films’ parameters were analyzed. It was observed that the VOC value slightly increases with the bulkiness of the side chains: the PE51, PE52, and PE53 polymers containing 2-butyloctyl, 2-hexyldecyl, and 2-octyldodecyl chains, respectively, exhibited VOC values of 0.79, 0.80, and 0.83 V, respectively. The authors attributed these differences to the changes in HOMO levels induced by the side chains. The trend was different if considering the molecular packing behavior investigated by GIWAXS: while PE51:Y6 and PE52:Y6 blend films exhibited an obvious (010) peak in the out-of-plane (OOP) direction, revealing a face-on orientation, the disappearance of this peak for PE53:Y6 blend film indicated that the polymer with the bulkiest side chains tends to stack out of the film plane. The calculated π–π stacking spacing for PE51:Y6, PE52:Y6, and PE53:Y6 (3.58, 3.48, and 3.73, respectively) and crystal coherence length (CCL) (25.41, 28.71, and 12.4 Å, respectively) indicated a strongest stacking, shortest charge-transport distance, and highest crystallinity for the PE52:Y6 blend. Consistently, it achieved the best JSC (25.36 mA cm−2) and FF (71.94%) values and reached the highest PCE of 14.61% among the examined series. These results were consistent with AFM and TEM analyses which revealed for the PE52:Y6 blend film an increased crystallization tendency of copolymer grains and the best quality of the bicontinuous interpenetrating network. Here, the best polymer is that with intermediate length of branched chains.
-
While the preceding examples relate to studies on polymer–small molecules systems, the next three deal with recent studies on all-polymer OPV cells. In the first case, three different D polymers P10 to P12 were investigated, with backbones all composed of alternating DTBDT, n-octyl-substituted thiophene, and thiazolothiazole (TTz) fragments (see Figure 1) [25]. The only difference between them was the nature of the alkyl chain introduced onto the pendant thiophene rings of the DTBDT fragment. The total number of carbon atoms introduced onto each thiophene ring was always the same, but the nature and number of chains differed from one polymer to another: two n-hexyl linear chains for DC6, one 2-butyloctyl branched chain for 2BO, and one n-dodecyl linear chain for C12. Absorption spectroscopy performed on thin films revealed no radical difference between them, which was not the case in chlorobenzene solutions in which DC6 showed a strong ability to form π-aggregates with increasing temperature unlike to the two other polymers. Optimized blends and solar cells have been developed using N2200 as the acceptor polymer. Their photovoltaic parameters revealed a clear difference between DC6, for which a PCE of 8.3% was obtained with a VOC of 0.87 V, JSC of 14.2 mA·cm−2, and FF of 66.4%, and the two other polymers for which the PCE value did not exceed 2.4%, due to low VOC and JSC values. These findings were explained by GIWAXS measurement which demonstrated weak face-on orientation of the backbone for 2BO and C12 while DC6 formed a highly ordered and strong face-on backbone stacking. Measurements of photocurrent vs. the applied voltage confirmed that DC6-based solar cells achieved the highest rate of exciton generation, and the dependencies of JSC and VOC on incident light intensity revealed negligible bimolecular recombination for DC6 containing two n-hexyl pendant chains per DTBDT unit unlike the other two polymers holding only one bulky linear or branched alkyl chain. Two hexyl chains were revealed to be the best option in that case.
-
In the next example, the effect of the alkyl chains was examined in an acceptor polymer, whose monomer unit was based on a seven-fused-ring system with a central benzotriazole core (see Figure 1, P13, P14) [26]. Butyloctyl or n-undecyl chains were introduced onto the polymer backbone whereas the N atom of the benzotriazole was substituted either by ethylhexyl or by a methyl group. Interestingly, the polymer with butyloctyl/ethylhexyl chains, PTz-BO, exhibited a higher absorption coefficient in solution but a narrower absorption range in thin film than that with n-undecyl/methyl chains, PTz-C11. After blending with PBDB-T as the donor polymer, the average electron/hole mobilities of PBDB-T:PTz-BO and PBDB-T:PTz-C11 were 3.34 × 10−4/2.26 × 10−4 and 3.71 × 10−4/2.22 × 10−4 cm2 V−1 s−1, with a slightly better μe/μh ratio for PBDB-T:PTz-BO. The PTz-BO-based device yielded better VOC and PCE values of 0.928 V and 15.76%, respectively, compared to the PTz-C11-based device (0.882 V and 15.59%), demonstrating a slightly positive effect of branched chains.
-
The third case is about an alternative method to tune the aggregation of alkylated polymer: random copolymerization. This was proposed by taking a naphthalene diimide (NDI)-based series of acceptor polymers (see Figure 1, P15) [23]. Random copolymerization between three monomers, namely 2,2′-bithiophene, and two 2,6-dibromonaphthalene-1,4,5,8-tetracarboxylic-diimide units substituted on their N,N’-position by either 2,6-diisopropyl-phenyl or 2-octyldodecyl fragments, respectively, afforded a series of acceptor materials with increasing ratio of aromatic side chain to branched alkyl chain (x = 0–1). After blending with the PBDB-T donor polymer, the nine all-polymer BHJs were submitted to morphology and photovoltaic characterization that emphasized the correlation of the optimal obtained PCE of 6.95% obtained by as-cast blends for x = 0.5 (vs. 5.11% and 1.06% for x = 0 and 1, respectively) with optimal active layer film morphology.
4. Ethoxy Chains
-
Another way to explore the effect of bulkiness of side chains was to use n-hexyloxy (C6) and oligoether (tri(ethyleneoxy) (EO)) chains grafted on the meta only or on the meta- and para-positions of the peripheral phenyl ring of the quinoxaline moiety in thiophene–quinoxaline copolymers (see P16–P18 in Figure 1) [27]. The absorption spectra of the copolymers with meta-para-disubstitution on the phenyl rings (TQ-4OC6 and TQ-4EO) are blue shifted in comparison with that of the meta-substituted copolymer (TQ-2OC6). This was attributed to smaller effective conjugation lengths, as suggested by density functional theory (DFT) results: the calculations highlighted the formation of oligomer chains in a circular-type configuration in the case of meta-para-disubstitution, producing more or less extended chains, which is unfavorable to the formation of polymers of long conjugation length. Consequently, only modest OPV parameters were obtained after blending with PC61BM, with PCEs not exceeding 1.27% for the meta-para-disubstituted species and 5.70% for the meta-monosubstituted one, after blending with PC61BM as the acceptor. This was surprising owing to PCE of 7% previously obtained with an analogous TQ blended with PC71BM and containing an n-octyl chain in the meta-position of the phenyl rings [28]. This may be related to the nature of the acceptor, although the deviation of the OPV parameters measured from one laboratory to another cannot be excluded.
-
The following example demonstrates the possible positive effect of ethoxy chain: two studies on copolymers based on fluorinated DTBDT and quinoxaline units (see P19, P20, Figure 1) were carried out [29][30]. The quinoxaline unit was dialkyl-substituted in PBQ5 and monoalkoxy-substituted in PBQ10. In the two cases, blends with Y6 as the acceptor were elaborated. The best photovoltaic results obtained with PBQ10 (PCE of 16.34% vs. 15.55% for PBQ5) cannot be related to difference in energy offset of the donor/acceptor as the two polymers presented similar HOMO/LUMO energy levels. Hole mobilities, measured by the SCLC method, were of 2.2 × 10−4 and 5.22 × 10−4 cm2 V−1 s−1 for PBQ5 and PBQ10, respectively. For the PBQ5:Y6 blend film, the μh/μe ratio was 0.86. No number was reported for PBQ10:Y6 films. Owing to GIWAXS results, both neat polymers films showed preferential face-on molecular orientation with well-defined π−π stacking diffraction peaks ((010) peak) in the out-of-plane (OOP) direction. Well-defined diffraction peaks indicated π−π stacking distance of 3.72 and 3.68 Å for PBQ5 and PBQ10, respectively. Contrarily to PBQ5, PBQ10 also exhibited a lamellar distance of 21.12 Å in the OOP direction and a CCL of 27.1Å, indicating a better molecular self-assembly. In this example, the use of a single branched substituent in the alkoxy form is preferable to that of two ethylhexyl substituents.
5. Alkylsilyl Chains
Since its first report in 2008 [31], the BDT unit has often been used as a donor unit for polymer elaboration; in particular, it gave rise to its derivative, DTBDT, well known as a donor moiety in A-D-A materials. Similar donor building blocks are widely used because their symmetrical chemical structures and rigid fused aromatic systems benefit the electron delocalization and π–π stacking [5] and contributed until very recently to the achievement of outstanding OPV device performances [32]. The DTBDT moiety is built from the commercially available benzo [1,2-b:4,5-b′]dithiophene-4,8-dione and offers the possibility of introducing a large variety of side chains on the pendant thiophene units. In particular, it has contributed to the generation of PBDB-T-based polymers (see P36, Figure 1), a significant milestone in OPVs, which afforded PCEs up to 11.14% in 2019 for a large-scale blade coated binary system [33] and up to 15.83% in 2022 at the laboratory scale [34]. Some specific DTBDT-based polymers (P21 to P32), designed for investigating the effect of bulky silyl groups, will be discussed hereafter.
-
In 2018, Bin and collaborators examined in a systematic manner ((DTBDT/benzotriazole)-based copolymers (see Figure 1, P21–P25) [35]. This polymer series was synthesized for investigating the length and linear or branched character of the alkyl substituents of alkylsilyl side chains of the DTBDT moiety. The optical gaps of the five polymers in thin films were very similar, from 1.96 to 1.99 eV. However, the electrochemical gap changed from 2.13 eV (J72) to 2.50 eV (J74), due to a slightly higher oxidation potential when the bulkiness increased from J70 to J74 together with slightly different values of reduction potential: J70 and J73 were reduced at about −1.45 V/Ag/AgCl, J71 and J72 at about −1.10 V/Ag/AgCl, with J74 being in between. GIWAXS analysis showed that the polymers in neat films are predominantly face-on oriented. The out-of-plane (OOP) π–π stacking distance gradually increased from 3.735 Å for J70, to 3.778 Å for J71, 3.792 Å for J72, 3.859 Å for J73, and 3.862 Å for J74, resulting from the effect of bulkiness of the alkylsilyl side chains. Despite modest electron and hole mobility values (below 10−3 cm2 V−1 s−1), good photovoltaic performances were obtained from solar cells with m-ITIC as the acceptor, exhibiting PCEs ranging from 9.63% (J74, the polymer with the longer chains) to 12.05% and 11.62% (for J71 and J70, respectively, the polymers with the shorter chains).
-
At the same time, the J47 polymer was engineered on the basis of the structure of J71 but with pendant thienothiophenyl fragments attached to the benzodithiophene moiety as an alternative to the thienyl ones (see Figure 1, P27) [36]. This polymer was compared to J46 presenting ethylhexyl instead of tripropylsilyl substituents (see Figure 1, P26). The photovoltaic performances of solar cells elaborated with (3,9-bis(2methylene-(3-(1,1-dicyanomethylene)indanone))-5,5,11,11-tetrakis(4-n-hexylphenyl)-dithieno [2,3d:2′,3′d’]-s-indaceno [1,2b:5,6b′]dithiophene) (ITIC) as the acceptor displayed a maximum PCE of 2.3% for the J46-based system and 7.4% for the J47-based solar cell. This difference was related to the JSC values that were two times lower for J46 (7.39 mA cm−2 vs. 16.22 mA cm−2 for J47) explained by the authors by the low hole mobility and unbalanced charge carrier mobilities in the J46:ITIC blend (μh = 0.40 × 10−5 cm2 V−1 s−1; μe/μh = 27.5) as compared to the J47:ITIC blend (μh = 1.85 × 10−4 cm2 V−1 s−1; μe/μh = 2.25).
-
Keeping the triisopropylsilyl substituent on the DTBDT moiety copolymerized with 1,3-bis(thiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo-[1,2-c:4,5-c’]dithiophene-4,8-dione (BDD), Huang and collaborators obtained the PBDS-T polymer (see Figure 1, P29)). They compared it to its homologue PBDB-T with ethylhexyl branched chains (P28) [37]. UV-Vis absorption spectrometry showed that PBDS-T had a higher absorption coefficient than PBDB-T and a better crystallinity, as confirmed by low-angle X-ray diffraction. Cyclic voltammetry revealed lower HOMO and LUMO energy levels for this polymer, as suggested by density functional theory (DFT) calculations. This was attributed to σ*(Si)–π*(C) bond interaction leading to an effective reduction in the energy levels of the silylated polymer. Moreover, steady-state and transient photoluminescence measurements demonstrated that PBDS-T:ITIC films present an improved crystallinity relative to that of PBDB-T:ITIC. This was consistent with better photovoltaic performances for PBDS-T:ITIC that led to a PCE of 11.06% with a VOC of 0.985 V, a JSC of 18.68 mA cm−2, and an FF of 60% in comparison to those of PBDB-T:ITIC (PCE of 9.80% with a VOC of 0.869 V, a JSC of 16.58 mA cm−2, and an FF of 68%). Later on, the same authors reached higher photovoltaic performances by improving the active layer morphology through a subtle variation of the NFA structure (PCE of 12.04% with a VOC of 0.88 V, a JSC of 19.42 mA cm−2, and an FF of 70%) [38]. The PBDS-T polymer subsequently used in combination with BTP-eC9 as the acceptor led to solar cells with further improved PCE of 16.4% [39], here also explained by enhanced film crystallinity and nanostructured packing of PBDS-T: BTP-eC9 blends. Polymers J71, J47, and PBDS-T all had in common that they were substituted by the same tripropylsilyl fragment.
-
Another polymer containing this fragment, named as PE6 (see Figure 1, P30), with a structure close to that of J71, recently displayed a high-power conversion efficiency of 15.09% by blending with 2,2′-((2Z,2′Z)-(12,13-bis(2ethylhexyl)-3,9-diundecyl-12,13-dihydro-[40][41][42]thiadiazolo [3,4e]thieno [2,”3”:4′,5′]thieno [2”,3”:4,5]pyrrolo [3,2-g]thieno [2′,3′:4,5]thieno [3,2-b]indole-2,1′-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene)dimalononitrile) (Y6) as the acceptor [43]. Recently, Genene and collaborators compared the two polymers PBDTTSi-TzBI and PBDTTS-TzBI, based on DTBDT and containing alkylsilyl and alkylthio side chains, respectively, associated with an acceptor imide-fused benzotriazoles (TzBI) acceptor block, itself substituted by two invariant alkyl (ethylhexyl and octyl) chains (see Figure 1, P31, P32) [44]. Electrochemical characterization highlighted the higher propensity of the trialkylsilyl side chains to lower the HOMO level as compared to the thioalkyl ones: the HOMO/LUMO energy levels were −5.46/−3.33 and −5.38/−3.32 eV for PBDTTSi-TzBI and PBDTTS-TzBI, respectively. However, PBDTTS-TzBI-based devices yielded better PCE values (7.32% and 9.60%) than PBDTTSi-TzBI systems (3.98% and 6.85%), using PC71BM or ITIC as the acceptor, respectively. No particular different features were denoted by AFM or TEM examination. The best hole/electron mobilities for PBDTTS-TzBI:ITIC and PBDTTSi-TzBI:ITIC were 9.27 × 10−4/7.32 × 10−4 cm2 V−1 s−1 and 5.24 × 10−4/1.98 × 10−4 cm2 V−1 s−1, respectively, showing more balanced μh/μe charge carrier mobilities for the alkylthio-based polymer. No specific investigations were reported on the even–odd effect of alkyl chains substituting the silicon atom on thin film structure and morphology as has been shown for alkyl-substituted small molecules [45].
6. Alkylthio Chains
-
As seen just above, the introduction of a sulfur atom between the thiophene lateral moiety of the donor moiety DTBDT and an alkyl chain may have a positive effect on photovoltaic performances. This has been the subject of a specific investigation for a DTBDT-based material copolymerized with fluorinated benzothiadiazole and thiophene units (see P33–P35, Figure 1) [46]. The thiophene rings attached to the BDT core were substituted in the 4-position by a bulky n-decyl chain and, and in the 5-position, either by another n-decyl linear chain for P33, by an n-thiodecyl chain for P34, or by a branched 2-ethylhexyl chain for P35. The first observation was a lower solubility of P33 in 1,2-dichlorobenzene in comparison to the two other polymers. No drastic difference was observed between the optical band gaps of the three polymers (1.63 to 1.67 eV). P34 and P35 displayed similar electrochemical behaviors, but with a HOMO energy of about −5.7 eV, i.e., 0.2 eV lower than that of P33, as the signature of the electronic impact of the presence of sulfur atoms. Once blended with PC71BM as the acceptor, the best performing polymer in the OPV device was P35, with a PCE of 9.0%, compared to 7.9 and 6.8% for P34 and P33, respectively, in agreement with the better balanced μe and μh values obtained for P35. This was explained by a difference in blend morphology revealed by AFM analysis. No phase contrast between the donor and acceptor phases was observed in P33:PC71BM films which was a sign of high miscibility of the two materials, contrarily to P34:PC71BM blend films that exhibited an average domain size of 80–90 nm of segregated phases of fullerene and polymer, responsible for charge recombination. P35:PC71BM films displayed a surface morphology compatible with an interpenetrating network well suited for generation and separation of charges, demonstrated here the advantage of using a branched chain over the thiodecyl one as the pendant thiophene substituent.
-
In that spirit, Chang and collaborators proposed to test thioalkyl pendant chains for tuning the phase crystallinity of a blend and, thus, its morphology [47]. Two donor polymers were investigated, PBDB-TF used as the host polymer and PBDB-T-SF used as the guest polymer (see P36, P37, Figure 1). Note that PBDB-TF is also known as PBDB-T-2F or more commonly as PM6, a very performant polymer that recently led to high PCE values on a large-area flexible module (up to 13.2% for 54 cm2) [48]. PBDB-TF and PBDB-T-SF presented identical backbone structures but different side chains grafted on the 5-position of the pendant thienyl rings of the DTBDT moiety, ethylhexyl for PBDB-T and thioethylhexyl for PBDB-TSF. This resulted in a difference in orientation of the side chains at the 4,8-positions of the BDT moiety that contributed to lowering the phase crystallinity of the host polymer by loading the guest polymer. Blends of polymers were associated with Y6 as the acceptor. By fine-tuning the PBDB-TF:PBDB-T-SF ratio, the authors demonstrated that it was possible to optimize the original fibrils’ size, from 10–20 nm and 30–60 nm for PBDB-TF:Y6 and PBDB-T-SF:Y6 binary blends, respectively, to over 100 nm for the for PBDB-TF:PBDB-T-SF:Y6 ternary blend. This meso-scale morphology was favorable to increased and well-balanced electron and hole mobilities, leading to an improved PCE of 16.42% as compared to the PCE of 15.50% obtained for the original PBDB-TF:Y6 binary blend.
7. Influence of the Length and Linear/Branched Nature of Carbonyl Side Chain
Introducing a carbonyl function between the conjugated backbone of a polymer and an alkyl side chain is also a way to modify intermolecular interactions and thus aggregation properties of conjugated polymers [49][50]. Polymers presenting twisted conjugated skeletons, based on difluorinated DTBDT and terthiophene, have been investigated. The terthiophene unit was substituted on the 3- and 3”-positions by either an n-ethylhexyl chain (PBDT-3T), a 2-ethylhexyl ketone (PBDT-3TCO), an n-decyl ketone fragment (P3TCO-D), or a 3,7-dimethyloctyl ketone (P3TCO-DMO) as depicted in Figure 1 (P38–P41). Photovoltaic performances were evaluated for blends with IT-4F as the acceptor. A PBDT-3T:IT-4F-based device gave a markedly lower PCE value (1.05%) than the others (10.1 to 12.8%), that was attributed to the quality of their respective BHJ morphologies. The key parameter identified was the introduction of the function (C=O) in the structure of the polymer which led to intra/intermolecular interactions of S···O and O···H reinforcing aggregation capacity between the polymer chains and the use of an appropriate bulky chain which modulated this aggregation. Branched chains of 3,7-dimethyloctyl have been shown to be best at tuning bicontinuous phase separation and improving charge carriers’ mobilities, purity, and domain size.
References
- Skotheim, T.A. (Ed.) Handbook of Conducting Polymers, Volume 1; Marcel Dekker: New York, NY, USA, 1986.
- Schopp, N.; Brus, V.V. A Review on the Materials Science and Device Physics of Semitransparent Organic Photovoltaics. Energies 2022, 15, 4639.
- Zhang, X.; Huang, H. Polymer-Related Organic Photovoltaics. Macromol. Rapid Commun. 2022, 43, 2200770.
- Al-Azzawi, A.G.S.; Aziz, S.B.; Dannoun, E.M.A.; Iraqi, A.; Nofal, M.M.; Murad, A.R.; Hussein, A.M. A Mini Review on the Development of Conjugated Polymers: Steps towards the Commercialization of Organic Solar Cells. Polymers 2023, 15, 164.
- Qiu, B.; Lai, J.; Yuan, J.; Zou, Y.; Li, Y. Recent Research Progress in Random Copolymerization of Polymer Photovoltaic Materials for High-Performance Polymer Solar Cells. Sol. RRL 2023, 7, 2300146.
- Tetreault, A.R.; Dang, M.-T.; Bender, T.P. PTB7 and PTB7-Th as Universal Polymers to Evaluate Materials Development Aspects of Organic Solar Cells Including Interfacial Layers, New Fullerenes, and Non-Fullerene Electron Acceptors. Synth. Met. 2022, 287, 117088.
- Cong, P.; Wang, Z.; Geng, Y.; Meng, Y.; Meng, C.; Chen, L.; Tang, A.; Zhou, E. Benzothiadiazole-Based Polymer Donors. Nano Energy 2023, 105, 108017.
- An, C.; Hou, J. BenzoDithiophene-Based Conjugated Polymers for Highly Efficient Organic Photovoltaics. Acc. Mater. Res. 2022, 3, 540–551.
- Hacefendioglu, T.; Yildirim, E. Design Principles for the Acceptor Units in Donor-Acceptor Conjugated Polymers. ACS Omega 2022, 7, 38969–38978.
- Soonmin, H.; Hardani; Nandi, P.; Mwankemwa, B.S.; Malevu, T.D.; Malik, M.I. Overview on Different Types of Solar Cells: An Update. Appl. Sci. 2023, 13, 2051.
- Guo, Q.; Guo, Q.; Geng, Y.; Tang, A.; Zhang, M.; Du, M.; Sun, X.; Zhou, E. Recent Advances in PM6:Y6-Based Organic Solar Cells. Mater. Chem. Front. 2021, 5, 3257–3280.
- Kim, M.; Choi, Y.; Hwan Lee, D.; Min, J.; Pu, Y.-J.; Park, T. Roles and Impacts of Ancillary Materials for Multi-Component Blend Organic Photovoltaics towards High Efficiency and Stability. ChemSusChem 2021, 14, 3475–3487.
- Meng, D.; Zheng, R.; Zhao, Y.; Zhang, E.; Dou, L.; Yang, Y. Near-Infrared Materials: The Turning Point of Organic Photovoltaics. Adv. Mater. 2022, 34, 2107330.
- Wenderott, J.K.; Dong, B.X.; Green, P.F. Morphological Design Strategies to Tailor Out-of-Plane Charge Transport in Conjugated Polymer Systems for Device Applications. Phys. Chem. Chem. Phys. 2021, 23, 27076–27102.
- Liu, Q.; Jiang, Y.; Jin, K.; Qin, J.; Xu, J.; Li, W.; Xiong, J.; Liu, J.; Xiao, Z.; Sun, K.; et al. 18% Efficiency Organic Solar Cells. Sci. Bull. 2020, 65, 272–275.
- Zhu, L.; Zhang, M.; Xu, J.; Li, C.; Yan, J.; Zhou, G.; Zhong, W.; Hao, T.; Song, J.; Xue, X.; et al. Single-Junction Organic Solar Cells with over 19% Efficiency Enabled by a Refined Double-Fibril Network Morphology. Nat. Mater. 2022, 21, 656–663.
- Mei, J.; Bao, Z. Side Chain Engineering in Solution-Processable Conjugated Polymers. Chem. Mater. 2014, 26, 604–615.
- Weng, K.; Ye, L.; Zhu, L.; Xu, J.; Zhou, J.; Feng, X.; Lu, G.; Tan, S.; Liu, F.; Sun, Y. Optimized Active Layer Morphology toward Efficient and Polymer Batch Insensitive Organic Solar Cells. Nat. Commun. 2020, 11, 2855.
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151.
- Li, N.; Perea, J.D.; Kassar, T.; Richter, M.; Heumueller, T.; Matt, G.J.; Hou, Y.; Güldal, N.S.; Chen, H.; Chen, S.; et al. Abnormal Strong Burn-in Degradation of Highly Efficient Polymer Solar Cells Caused by Spinodal Donor-Acceptor Demixing. Nat. Commun. 2017, 8, 14541.
- Tegegne, N.A.; Abdissa, Z.; Mammo, W.; Uchiyama, T.; Okada-Shudo, Y.; Galeotti, F.; Porzio, W.; Andersson, M.R.; Schlettwein, D.; Vohra, V.; et al. Effect of Alkyl Side Chain Length on Intra- and Intermolecular Interactions of Terthiophene-Isoindigo Copolymers. J. Phys. Chem. C 2020, 124, 9644–9655.
- Zhang, C.-H.; Lin, F.; Huang, W.; Xin, J.; Wang, J.; Lin, Z.; Ma, W.; Yang, T.; Xia, J.; Liang, Y. Methyl Functionalization on Conjugated Side Chains for Polymer Solar Cells Processed from Non-Chlorinated Solvents. J. Mater. Chem. C Mater. Opt. Electron. Devices 2020, 8, 11532–11539.
- Wu, Y.; Schneider, S.; Walter, C.; Chowdhury, A.H.; Bahrami, B.; Wu, H.-C.; Qiao, Q.; Toney, M.F.; Bao, Z. Fine-Tuning Semiconducting Polymer Self-Aggregation and Crystallinity Enables Optimal Morphology and High-Performance Printed All-Polymer Solar Cells. J. Am. Chem. Soc. 2020, 142, 392–406.
- Zhou, J.; Zhang, B.; Du, M.; Dai, T.; Tang, A.; Guo, Q.; Zhou, E. Side-Chain Engineering of Copolymers Based on Benzotriazole (BTA) and DithienoBenzoDithiophenes (DTBDT) Enables a High PCE of 14.6%. Nanotechnology 2021, 32, 225403.
- Chen, J.; Huang, X.; Cao, Z.; Liu, S.; Liang, K.; Liu, J.; Jiao, X.; Zhao, J.; Li, Q.; Cai, Y.-P. Pronounced Dependence of All-Polymer Solar Cells Photovoltaic Performance on the Alkyl Substituent Patterns in Large Bandgap Polymer Donors. ChemPhysChem 2020, 21, 908–915.
- Li, X.; Duan, X.; Qiao, J.; Li, S.; Cai, Y.; Zhang, J.; Zhang, Y.; Hao, X.; Sun, Y. Benzotriazole-Based Polymer Acceptor for High-Efficiency All-Polymer Solar Cells with High Photocurrent and Low Voltage Loss. Adv. Energy Mater. 2023, 13, 2203044.
- Costa, C.; Farinhas, J.; Galvao, A.M.; Charas, A. On the Effect of Pattern Substitution and Oligo(Ethylene Oxide) Side-Chain Modification on Thiophene-Quinoxaline Copolymers and Their Applications in Photovoltaic Cells. Org. Electron. 2020, 78, 105612.
- Kim, Y.; Yeom, H.R.; Kim, J.Y.; Yang, C. High-Efficiency Polymer Solar Cells with a Cost-Effective Quinoxaline Polymer through Nanoscale Morphology Control Induced by Practical Processing Additives. Energy Environ. Sci. 2013, 6, 1909.
- Sun, C.; Pan, F.; Qiu, B.; Qin, S.; Chen, S.; Shang, Z.; Meng, L.; Yang, C.; Li, Y. D-A Copolymer Donor Based on Bithienyl Benzodithiophene D-Unit and Monoalkoxy Bifluoroquinoxaline A-Unit for High-Performance Polymer Solar Cells. Chem. Mater. 2020, 32, 3254–3261.
- Zhu, C.; Meng, L.; Zhang, J.; Qin, S.; Lai, W.; Qiu, B.; Yuan, J.; Wan, Y.; Huang, W.; Li, Y. A Quinoxaline-Based D-A Copolymer Donor Achieving 17.62% Efficiency of Organic Solar Cells. Adv. Mater. 2021, 33, 2100474.
- Hou, J.; Park, M.-H.; Zhang, S.; Yao, Y.; Chen, L.-M.; Li, J.-H.; Yang, Y. Bandgap and Molecular Energy Level Control of Conjugated Polymer Photovoltaic Materials Based on BenzoDithiophene. Macromolecules 2008, 41, 6012–6018.
- Shang, A.; Luo, S.; Zhang, J.; Zhao, H.; Xia, X.; Pan, M.; Li, C.; Chen, Y.; Yi, J.; Lu, X.; et al. Over 18% Binary Organic Solar Cells Enabled by Isomerization of Non-Fullerene Acceptors with Alkylthiophene Side Chains. Sci. China Chem. 2022, 65, 1758–1766.
- Sun, L.; Fukuda, K.; Someya, T. Recent Progress in Solution-Processed Flexible Organic Photovoltaics. Npj Flex. Electron. 2022, 6, 89.
- Busireddy, M.R.; Chen, T.-W.; Huang, S.-C.; Su, Y.-J.; Wang, Y.-M.; Chuang, W.-T.; Chen, J.-T.; Hsu, C.-S. PBDB-T-Based Binary-OSCs Achieving over 15.83% Efficiency via End-Group Functionalization and Alkyl-Chain Engineering of Quinoxaline-Containing Non-Fullerene Acceptors. ACS Appl. Mater. Interfaces 2022, 14, 41264–41274.
- Bin, H.; Yang, Y.; Peng, Z.; Ye, L.; Yao, J.; Zhong, L.; Sun, C.; Gao, L.; Huang, H.; Li, X.; et al. Effect of Alkylsilyl Side-Chain Structure on Photovoltaic Properties of Conjugated Polymer Donors. Adv. Energy Mater. 2018, 8, 1702324.
- Yan, T.; Bin, H.; Sun, C.; Zhang, Z.-G.; Li, Y. Synthesis and Photovoltaic Properties of 2D-Conjugated Polymers with Alkylsilyl-Substituted ThienoThiophene Conjugated Side Chains. Org. Electron. 2018, 57, 255–262.
- Huang, B.; Chen, L.; Jin, X.; Chen, D.; An, Y.; Xie, Q.; Tan, Y.; Lei, H.; Chen, Y. Alkylsilyl Functionalized Copolymer Donor for Annealing-Free High Performance Solar Cells with over 11% Efficiency: Crystallinity Induced Small Driving Force. Adv. Funct. Mater. 2018, 28, 1800606.
- Huang, B.; Hu, L.; Chen, L.; Chen, S.; Hu, M.; Zhou, Y.; Zhang, Y.; Yang, C.; Chen, Y. Morphological Optimization by Rational Matching of the Donor and Acceptor Boosts the Efficiency of Alkylsilyl Fused Ring-Based Polymer Solar Cells. J. Mater. Chem. Mater. Energy Sustain. 2019, 7, 4847–4854.
- Bin, H.; Van Der Pol, T.P.A.; Li, J.; Van Gorkom, B.T.; Wienk, M.M.; Janssen, R.A.J. Efficient Organic Solar Cells with Small Energy Losses Based on a Wide-Bandgap Trialkylsilyl-Substituted Donor Polymer and a Non-Fullerene Acceptor. Chem. Eng. J. 2022, 435, 134878.
- Solak, E.K.; Irmak, E. Advances in Organic Photovoltaic Cells: A Comprehensive Review of Materials, Technologies, and Performance. RSC Adv. 2023, 13, 12244–12269.
- Zhou, Y.; Luo, X.; Yang, J.; Qiu, Q.; Xie, T.; Liang, T. Application of Quantum Dot Interface Modification Layer in Perovskite Solar Cells: Progress and Perspectives. Nanomaterials 2022, 12, 2102.
- Li, F.; Xie, Y.; Hu, Y.; Long, M.; Zhang, Y.; Xu, J.; Qin, M.; Lu, X.; Liu, M. Effects of Alkyl Chain Length on Crystal Growth and Oxidation Process of Two-Dimensional Tin Halide Perovskites. ACS Energy Lett. 2020, 5, 1422–1429.
- Zhou, J.; Lei, P.; Geng, Y.; He, Z.; Li, X.; Zeng, Q.; Tang, A.; Zhou, E. A Linear 2D-Conjugated Polymer Based on 4,8-Bis(4-Chloro-5-Tripropylsilyl-Thiophen-2-Yl)BenzoDithiophene (BDT-T-SiCl) for Low Voltage Loss Organic Photovoltaics. J. Mater. Chem. A 2022, 10, 9869–9877.
- Genene, Z.; Negash, A.; Abdulahi, B.A.; Eachambadi, R.T.; Liu, Z.; Van den Brande, N.; D’Haen, J.; Wang, E.; Vandewal, K.; Maes, W.; et al. Comparative Study on the Effects of Alkylsilyl and Alkylthio Side Chains on the Performance of Fullerene and Non-Fullerene Polymer Solar Cells. Org. Electron. 2020, 77, 105572.
- Akkerman, H.B.; Mannsfeld, S.C.B.; Kaushik, A.P.; Verploegen, E.; Burnier, L.; Zoombelt, A.P.; Saathoff, J.D.; Hong, S.; Atahan-Evrenk, S.; Liu, X.; et al. Effects of Odd–Even Side Chain Length of Alkyl-Substituted Diphenylbithiophenes on First Monolayer Thin Film Packing Structure. J. Am. Chem. Soc. 2013, 135, 11006–11014.
- Kuznetsov, I.E.; Nikitenko, S.L.; Kuznetsov, P.M.; Dremova, N.N.; Troshin, P.A.; Akkuratov, A.V. Solubilizing Side Chain Engineering: Efficient Strategy to Improve the Photovoltaic Performance of Novel Benzodithiophene-Based (X-DADAD)n Conjugated Polymers. Macromol. Rapid Commun. 2020, 41, 2000430.
- Chang, Y.; Lau, T.-K.; Chow, P.C.Y.; Wu, N.; Su, D.; Zhang, W.; Meng, H.; Ma, C.; Liu, T.; Li, K.; et al. A 16.4% Efficiency Organic Photovoltaic Cell Enabled Using Two Donor Polymers with Their Side-Chains Oriented Differently by a Ternary Strategy. J. Mater. Chem. Mater. Energy Sustain. 2020, 8, 3676–3685.
- Qin, F.; Sun, L.; Chen, H.; Liu, Y.; Lu, X.; Wang, W.; Liu, T.; Dong, X.; Jiang, P.; Jiang, Y.; et al. 54 cm2 Large-Area Flexible Organic Solar Modules with Efficiency Above 13%. Adv. Mater. 2021, 33, 2103017.
- An, C.; Xin, J.; Shi, L.; Ma, W.; Zhang, J.; Yao, H.; Li, S.; Hou, J. Enhanced Intermolecular Interactions to Improve Twisted Polymer Photovoltaic Performance. Sci. China Chem. 2019, 62, 370–377.
- Lv, Q.; An, C.; Zhang, T.; Zhou, P.; Hou, J. Effect of Alkyl Side Chains of Twisted Conjugated Polymer Donors on Photovoltaic Performance. Polymer 2021, 218, 123475.
More