Efficient brain function requires as much as 20% of the total oxygen intake to support normal neuronal cell function. This level of oxygen usage, however, leads to the generation of free radicals, and thus can lead to oxidative stress and potentially to age-related cognitive decay and even neurodegenerative diseases. The regulation of this system requires a complex monitoring network to maintain proper oxygen homeostasis. Furthermore, the high content of mitochondria in the brain has elevated glucose demands, and thus requires a normal redox balance. Maintaining this is mediated by adaptive stress response pathways that permit cells to survive oxidative stress and to minimize cellular damage. These stress pathways rely on the proper function of the endoplasmic reticulum (ER) and the activation of the unfolded protein response (UPR), a cellular pathway responsible for normal ER function and cell survival.
- endoplasmic reticulum stress
- mitochondria unfolded protein response
- oxidative stress
- neurodegeneration
- proteostasis
- calcium
- brain
- nitrosative stress
- oxygen homeostasis
1. Introduction
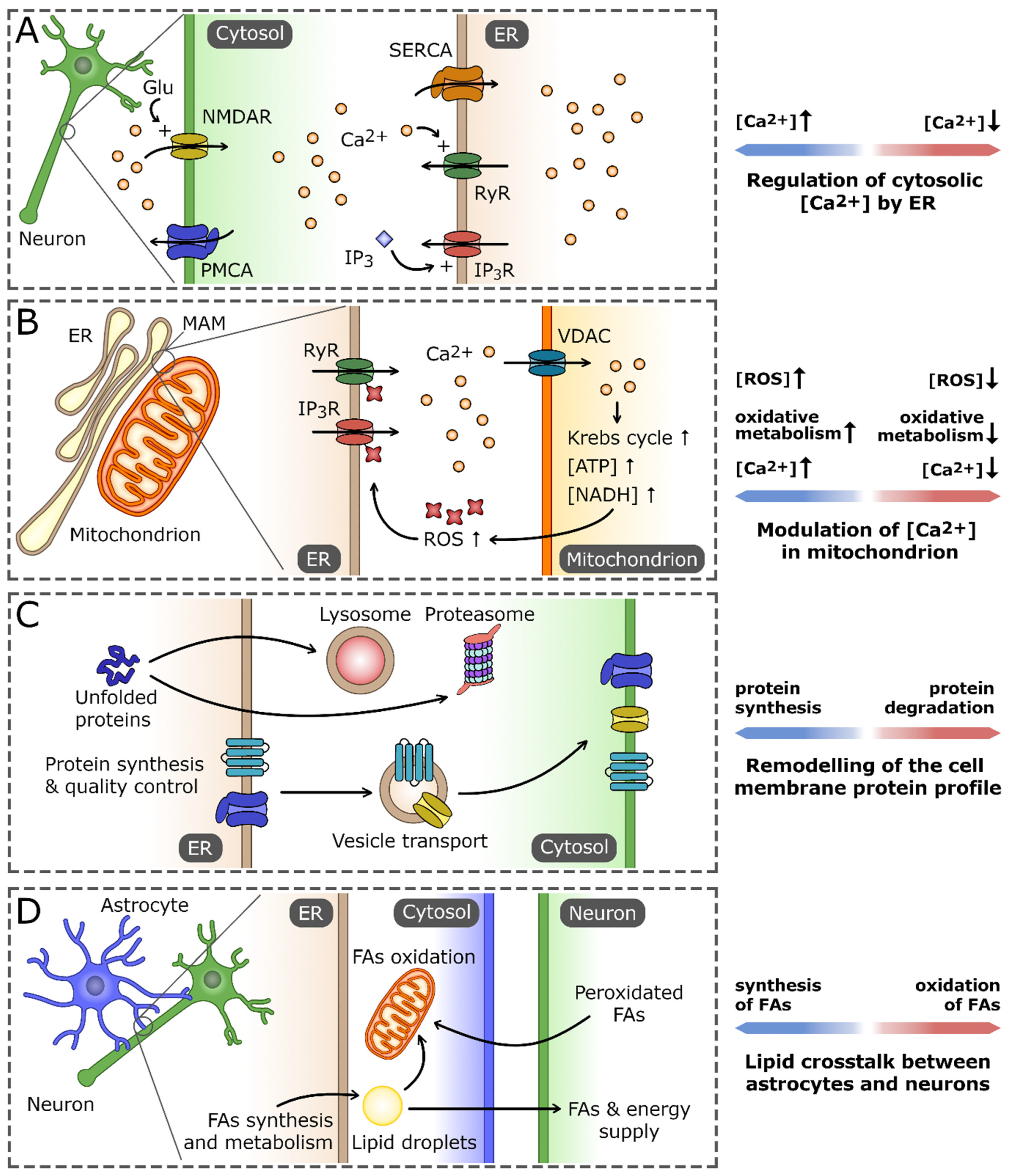
2. Role of the ER in Maintaining Neuron Cell Homeostasis
2.1. Calcium Regulation and Signaling
2.2. The ER and Proteostasis
2.3. The ER Lipid Biosynthesis
References
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212.
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57.
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530.
- Desai, S.; Rocha, M.; Jovin, T.; Jadhav, A. High Variability in Neuronal Loss: Time is Brain, Re-quantified. Stroke 2019, 50, 34–37.
- Bailey, D.M.; Bartsch, P.; Knauth, M.; Baumgartner, R.W. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: From the molecular to the morphological. Cell Mol. Life Sci. 2009, 66, 3583–3594.
- Carter, R. Oxygen: The Molecule that made the World. J. R. Soc. Med. 2003, 96, 46–47.
- Sawyer, D.T.; Valentine, J.S. How Super Is Superoxide. Acc. Chem. Res. 1981, 14, 393–400.
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286.
- Sies, H. Biochemistry of Oxidative Stress. Eur. J. Cancer Clin. 1987, 23, 1798.
- Campese, V.M.; Ye, S.H.; Zhong, H.Q. Reactive oxygen species (ROS) and central regulation of the sympathetic nervous system (SNS) activity. Hypertension 2002, 40, 382.
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623.
- Gutowicz, M. The influence of reactive oxygen species on the central nervous system. Postep. Hig. Med. Dosw. 2011, 65, 104–113.
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658.
- Michalska, P.; Leon, R. When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants 2020, 9, 740.
- Quinn, P.M.J.; Ambrosio, A.F.; Alves, C.H. Oxidative Stress, Neuroinflammation and Neurodegeneration: The Chicken, the Egg and the Dinosaur. Antioxidants 2022, 11, 1554.
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647.
- Zeevalk, G.D.; Bernard, L.P.; Song, C.; Gluck, M.; Ehrhart, J. Mitochondrial inhibition and oxidative stress: Reciprocating players in neurodegeneration. Antioxid. Redox Signal. 2005, 7, 1117–1139.
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10 (Suppl. S7), S18–S25.
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205.
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605.
- Bailey, D.M.; Willie, C.K.; Hoiland, R.L.; Bain, A.R.; MacLeod, D.B.; Santoro, M.A.; DeMasi, D.K.; Andrijanic, A.; Mijacika, T.; Barak, O.F.; et al. Surviving Without Oxygen: How Low Can the Human Brain Go? High Alt. Med. Biol. 2017, 18, 73–79.
- Zucker, R.S. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. NeuroBiol. 1999, 9, 305–313.
- Wheeler, D.B.; Randall, A.; Tsien, R.W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 1994, 264, 107–111.
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273.
- Collin, T.; Franconville, R.; Ehrlich, B.E.; Llano, I. Activation of metabotropic glutamate receptors induces periodic burst firing and concomitant cytosolic Ca2+ oscillations in cerebellar interneurons. J. Neurosci. 2009, 29, 9281–9291.
- Huser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343 Pt 2, 311–317.
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099.
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498.
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem. 2005, 280, 24888–24894.
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766.
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248.
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529 Pt 1, 37–47.
- Nichols, B.J.; Denton, R.M. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell Biochem. 1995, 149, 203–212.
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys Acta 2009, 1787, 1309–1316.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065.
- Breckwoldt, M.O.; Pfister, F.M.; Bradley, P.M.; Marinkovic, P.; Williams, P.R.; Brill, M.S.; Plomer, B.; Schmalz, A.; St Clair, D.K.; Naumann, R.; et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 2014, 20, 555–560.
- Vasquez, G.E.; Medinas, D.B.; Urra, H.; Hetz, C. Emerging roles of endoplasmic reticulum proteostasis in brain development. Cells Dev. 2022, 170, 203781.
- Cohen-Cory, S. The developing synapse: Construction and modulation of synaptic structures and circuits. Science 2002, 298, 770–776.
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546.
- Martinez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER Proteostasis Control of Neuronal Physiology and Synaptic Function. Trends Neurosci. 2018, 41, 610–624.
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94.
- Kennedy, M.J.; Hanus, C. Architecture and Dynamics of the Neuronal Secretory Network. Annu. Rev. Cell Dev. Biol. 2019, 35, 543–566.
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919.
- Hetz, C. Adapting the proteostasis capacity to sustain brain healthspan. Cell 2021, 184, 1545–1560.
- Sossin, W.S.; Costa-Mattioli, M. Translational Control in the Brain in Health and Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a032912.
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys Acta 2013, 1833, 2410–2424.
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108.
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. Embo. Rep. 2005, 6, 28–32.
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129812.
- Kozlov, G.; Gehring, K. Calnexin cycle-structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340.
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090.
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163.
- Poulsen, H.E.; Specht, E.; Broedbaek, K.; Henriksen, T.; Ellervik, C.; Mandrup-Poulsen, T.; Tonnesen, M.; Nielsen, P.E.; Andersen, H.U.; Weimann, A. RNA modifications by oxidation: A novel disease mechanism? Free Radic. Biol. Med. 2012, 52, 1353–1361.
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. USA 2007, 104, 66–71.
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964.
- Ding, Q.; Dimayuga, E.; Keller, J.N. Oxidative stress alters neuronal RNA- and protein-synthesis: Implications for neural viability. Free Radic. Res. 2007, 41, 903–910.
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849.
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491.
- Freeman, O.J.; Mallucci, G.R. The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 2016, 1648, 530–537.
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544.
- Rouser, G.; Galli, C.; Kritchevsky, G. Lipid Class Composition of Normal Human Brain and Variations in Metachromatic Leucodystrophy, Tay-Sachs, Niemann-Pick, Chronic Gaucher’s and Alzheimer’s Diseases. J. Am. Oil. Chem. Soc. 1965, 42, 404–410.
- Puchkov, D.; Haucke, V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell. Biol. 2013, 23, 493–503.
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell Neurosci. 2019, 13, 212.
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535 e1514.
- Camargo, N.; Brouwers, J.F.; Loos, M.; Gutmann, D.H.; Smit, A.B.; Verheijen, M.H. High-fat diet ameliorates neurological deficits caused by defective astrocyte lipid metabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4302–4315.
- Chen, J.; Zhang, X.; Kusumo, H.; Costa, L.G.; Guizzetti, M. Cholesterol efflux is differentially regulated in neurons and astrocytes: Implications for brain cholesterol homeostasis. Biochim. Biophys. Acta 2013, 1831, 263–275.
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921.
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452.
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249.
- Mallucci, G.R.; Klenerman, D.; Rubinsztein, D.C. Developing Therapies for Neurodegenerative Disorders: Insights from Protein Aggregation and Cellular Stress Responses. Annu. Rev. Cell Dev. Biol. 2020, 36, 165–189.
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666.
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304.