Ovarian cancer is one of the most common gynecologic cancers and has the highest mortality rate of any other cancer of the female reproductive system. Epithelial ovarian cancer (EOC) accounts for approximately 90% of all ovarian malignancies. The standard therapeutic strategy includes cytoreductive surgery accompanied by pre- or postoperative platinum-based chemotherapy. Nevertheless, up to 80% of the patients relapse within the following 12–18 months from the completion of the treatment and then receive first-line chemotherapy depending on platinum sensitivity. Mutations in BRCA1/2 genes are the most significant molecular aberrations in EOC and serve as prognostic and predictive biomarkers. Poly ADP-ribose polymerase (PARP) inhibitors exploit defects in the DNA repair pathway through synthetic lethality.
- ovarian cancer
- BRCA mutations
- PARP inhibitors
- immune checkpoint inhibitors
1. PARP Inhibitors
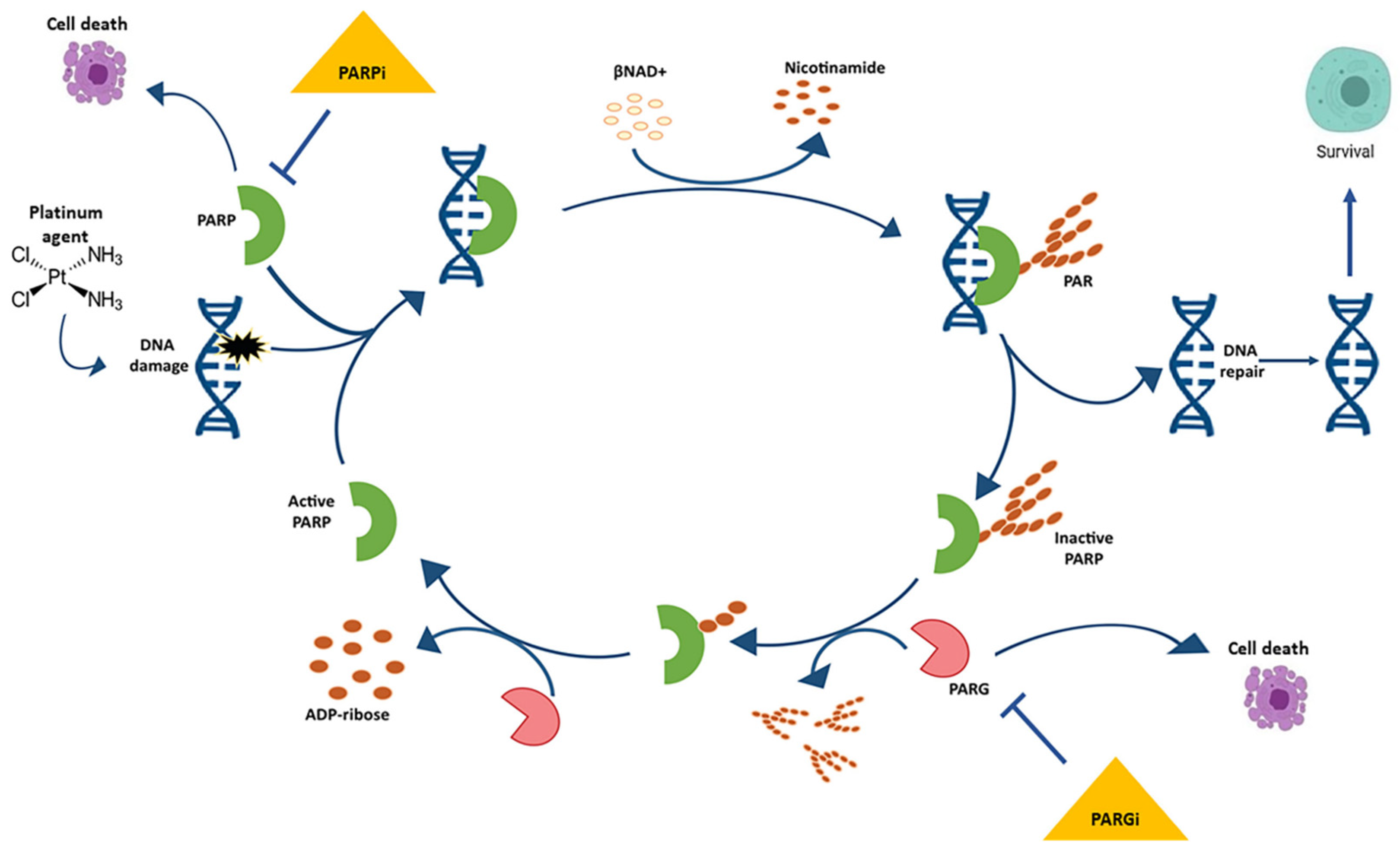
1.1. Olaparib
1.2. Niraparib
1.3. Rucaparib
1.4. Veliparib
1.5. Talazoparib
2. PARP Inhibitor Combination Therapies
| Combined Treatment (Type and Pathways) | Approval | Studies/Ref | Setting | Target | PARP Inhibitor |
|---|---|---|---|---|---|
| Antiangiogenics Immunotherapy PI3K/AKT/mTOR Wee1 Chemotherapy |
FDA and EMA | SOLO1, SOLO2, SOLO3/[6][35][36] | EOC with BRCA mutations HER2-negative BRCA-mutated breast cancer Metastatic pancreatic cancer with germline BRCA mutations Prostate cancer |
PARP-1, -2 and -3 | Olaparib |
| Antiangiogenics Immunotherapy |
FDA | ENGOTOV16/NOVA, PRIMA, QUADRA/[9][10][11] | Platinum-sensitive EOC with BRCA mutations | PARP-1 and -2 | Niraparib |
| Immunotherapy | FDA and EMA | ARIEL2, ARIEL 3, ARIEL 4/[14][15][16] | Monotherapy in advanced EOC with germline or somatic BRCA mutations | PARP-1, -2 and -3 and tankyrase-1 | Rucaparib |
| Chemotherapy Radiotherapy |
No | NCT01540565/[18] | EOC with BRCA mutations | PARP-1 and -2 | Veliparib |
| Immunotherapy Chemotherapy |
No | NCT01286987/[24] | HER2-negative breast cancer | PARP-1 and -2 | Talazoparib |
3. Mechanisms of PARP Inhibitors Resistance
References
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87.
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to Ithaca. Diagnostics 2019, 9, 55.
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663.
- FDA. Olaparib. Approvement. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/206162Orig1s000Approv.Pdf (accessed on 30 October 2017).
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861.
- Pujade-Lauraine, E.; Ledermann, J.A.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; Friedlander, M.; Colombo, N. Treatment with olaparib monotherapy in the maintenance setting significantly improves progression-free survival in patients with platinum sensitive relapsed ovarian cancer: Results from the phase III SOLO2 study. Gynecol. Oncol. 2017, 145, 219–220.
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019.
- Liu, J.F.; Konstantinopoulos, P.A.; Matulonis, U.A. PARP inhibitors in ovarian cancer: Current status and future promise. Gynecol. Oncol. 2014, 133, 362–369.
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164.
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402.
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648.
- Syed, Y.Y. Rucaparib: First global approval. Drugs 2017, 77, 585–592.
- FDA. Rucaparib. Approvement. FDA Grants Accelerated Approval to New Treatment for Advanced Ovarian Cancer|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-new-treatment-advanced-ovarian-cancer (accessed on 2 June 2022).
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87.
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961.
- Kristeleit, R.; Lisyanskaya, A.; Fedenko, A.; Dvorkin, M.; de Melo, A.C.; Shparyk, Y.; Rakhmatullina, I.; Bondarenko, I.; Colombo, N.; Svintsitskiy, V.; et al. Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): An international, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 465–478.
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737.
- Coleman, R.L.; Sill, M.W.; Bell-McGuinn, K.; Aghajanian, C.; Gray, H.J.; Tewari, K.S.; Rubin, S.C.; Rutherford, T.J.; Chan, J.K.; Chen, A.; et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation—An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2015, 137, 386–391.
- Kummar, S.; Oza, A.M.; Fleming, G.F.; Sullivan, D.M.; Gandara, D.R.; Naughton, M.J.; Villalona-Calero, M.A.; Morgan, R.J., Jr.; Szabo, P.M.; Youn, A.; et al. Randomized Trial of Oral Cyclophosphamide and Veliparib in High-Grade Serous Ovarian, Primary Peritoneal, or Fallopian Tube Cancers, or BRCA-Mutant Ovarian Cancer. Clin. Cancer Res. 2015, 21, 1574–1582.
- Veliparib with Carboplatin and Paclitaxel and as Continuation Maintenance Therapy in Subjects with Newly Diagnosed Stage III or IV, High-Grade Serous, Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02470585 (accessed on 2 June 2022).
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2020, 38, 181–193.
- Palma, J.P.; Wang, Y.C.; Rodriguez, L.E.; Montgomery, D.; Ellis, P.A.; Bukofzer, G.; Niquette, A.; Liu, X.; Shi, Y.; Lasko, L.; et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin. Cancer Res. 2009, 15, 7277–7290.
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015.
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, DoseEscalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629.
- Dréan, A.; Lord, C.J.; Ashworth, A. PARP inhibitor combination therapy. Crit. Rev. Oncol. Hematol. 2016, 108, 73–85.
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104.
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97.
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214.
- Matulonis, U.A.; Monk, B.J. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: Does a development pathway forward exist? Ann. Oncol. 2017, 28, 443–447.
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA damaging revolution: PARP inhibitors and beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195.
- Matulonis, U.A.; Wulf, G.; Barry, W.; Birrer, M.; Westin, S.; Spagnoletti, T.; Bell-McGuinn, K.; Obermayer, E.; Whalen, C.; Aghajanian, C.; et al. Phase I of oral BKM120 or BYL719 and olaparib for high-grade serous ovarian cancer or triple-negative breast cancer: Final results of the BKM120 plus olaparib cohort. Cancer Res. 2015, 75, CT324.
- Michalarea, V.; Lorente, D.; Lopez, J.; Carreira, S.; Hassam, H.; Parmar, M.; Sathiyayogan, N.; Turner, A.; Hall, E.; Fandos, S.S.; et al. Accelerated phase I trial of two schedules of the combination of the PARP inhibitor olaparib and AKT inhibitor AZD5363 using a novel intrapatient dose escalation design in advanced cancer patients. Cancer Res. 2015, 75, CT323.
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149.
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419.
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505.
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174.
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704.
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017, 7, 984–998.
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis-Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429.
- Lim, J.S.J.; Tan, D.S.P. Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers 2017, 9, 109.
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127.
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121.
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332.
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387.
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378.
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111.