Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Stergios Boussios.
Non-epithelial ovarian cancers (NEOC) are a group of uncommon malignancies that mainly includes germ cell tumours (GCT), sex cord-stromal tumours (SCST), and some extremely rare tumours, such as small cell carcinomas and sarcomas. Each of these classifications encompasses multiple histologic subtypes. The aetiology and molecular origins of each sub-group of NEOC require further investigation, and our understanding on the genetic changes should be optimised.
- germ cell tumours
- sex cord-stromal tumours
- small cell carcinomas of the ovary
1. Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynaecologic malignancy, as it is commonly diagnosed at an advanced stage. Non-epithelial ovarian cancers (NEOC) are rare, accounting for approximately 10% of all ovarian cancers and include mainly germ cell tumours (GCT), sex cord-stromal tumours (SCST), and some extremely rare tumours [1,2,3][1][2][3]. The greatest risk factors of ovarian cancer are a family history and associated genetic syndromes. Several modifiable risk factors, such as obesity, smoking and sedentary lifestyle, are associated with an increased risk of ovarian cancer, but have not been established as predisposition factors. Endometriosis is directly related to certain EOC subtypes, specifically clear cell and endometrioid ovarian carcinoma, rather than to NEOC [4].
GCT originate from the primordial germ cell and are divided into dysgerminomas and nondysgerminomas, including primarily yolk sac tumours (YST) and immature teratomas. GCT most often occur among younger women of childbearing age. However, cases of postmenopausal GCT, primarily YST, have been reported in the literature. SCST arise from the sex cord and ovarian stroma and comprise a heterogeneous group of tumours, of which granulosa cell tumour (GrCT) is the most frequent type. SCST are mainly diagnosed in older age groups with a median age of around 50 years. GrCT are divided into adult and juvenile types, of which the adult type represents approximately 95%. The Forkhead box L2 (FOXL2) gene mutation has been found to be an extremely sensitive and quite specific marker for the adult GrCT.
The initial symptoms and signs of NEOC are usually a subacute pelvic pain, feeling of pelvic pressure due of a pelvic mass and menstrual irregularities [2]. Symptoms become more noticeable as the cancer progresses. Diagnostic work-up should include pelvic ultrasound, an abdomino-pelvic computed tomography (CT) scan, chest X-ray and a positron emission tomography (PET) scan in selected cases. From the biochemical perspective, serum beta-hCG (β-hCG), α-foetoprotein (AFP) and lactate dehydrogenase (LDH) levels, along with liver and renal functions, should be carried out. Full blood count tests are also recommended. Inhibin B is secreted by GrCT and could be a useful marker for the disease. Serum anti-Müllerian hormone (AMH) may be a marker of ovarian reserve and GrCT in postmenopausal or post-oophorectomy women. While these markers are nonspecific, they can provide prognostic information, and as such quantitative hCG, AFP, LDH and cancer antigen 125 (CA-125) should be measured preoperatively [2]. Fertility-sparing surgery (FSS) and platinum-based chemotherapy remain the standard of care, providing a high chance of cure at all stages. Given the lack of high-quality studies in this field, current practice guidelines recommend chemotherapy regimens adopted in testicular GCT. However, platinum-resistant/refractory GCT retain a worse prognosis in comparison with their male counterpart.
2. Ovarian GCT
2.1. Dysgerminomas
Dysgerminomas are the most common GCT and can present at any age, but most of them occur in adolescence and early adulthood [15][5], with 85% of patients aged less than 30 years at time of diagnosis [16][6]. Patients will often present with abdominal pain, distension, and menstrual disorders [6][7]. There is nonspecific elevation of serum alkaline phosphatase (ALP) and lactic dehydrogenase, and 5% of dysgerminomas secrete β-hCG because they contain multinucleated syncytiotrophoblastic giant cells [6][7]. Dysgerminomas are solid and well encapsulated large masses, which on sectioning will reveal a soft, fleshy, and lobulated tan or grey surface [6][7]. Rarely would they cause haemorrhage or necrosis [10][8]. Histologically, uniform, large tumour cells are separated by fibrovascular septae which is almost always infiltrated with chronic inflammatory cells, especially lymphocytes [10,17][8][9]. This appearance is reflected on various modes of imaging. On ultrasound they will appear as smooth, well-defined lobules with heterogenous echogenicity and will be vascularised on colour and power Doppler ultrasound [18][10]. Furthermore, on CT and magnetic resonance imaging (MRI), a solid tumour will be seen with the fibrovascular septa along with areas of necrosis or haemorrhage if present [6,17][7][9]. Immunohistochemistry staining may also be positive for markers, such as placental alkaline phosphatase (PLAP), cluster of differentiation 117 (CD117) and D2-40, with c-KIT mutation being present in 33–50% of patients [10,19][8][11]. Dysgerminomas have a favourable prognosis, with a five-year survival of approximately 90%; this decreases to 63% if the disease has extended beyond the ovaries [10,20][8][12]. Recurrence rates may range between 18–52%, with over 75% of these occurring in the first year after diagnosis [7,10][8][13].
2.2. Yolk Sac Tumours
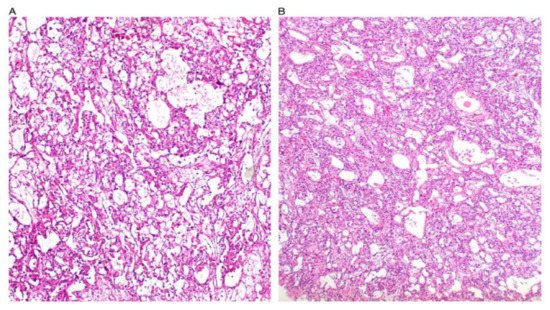
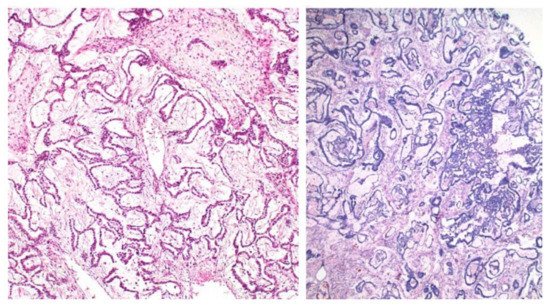
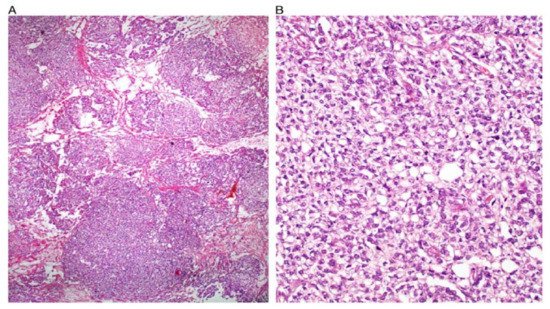
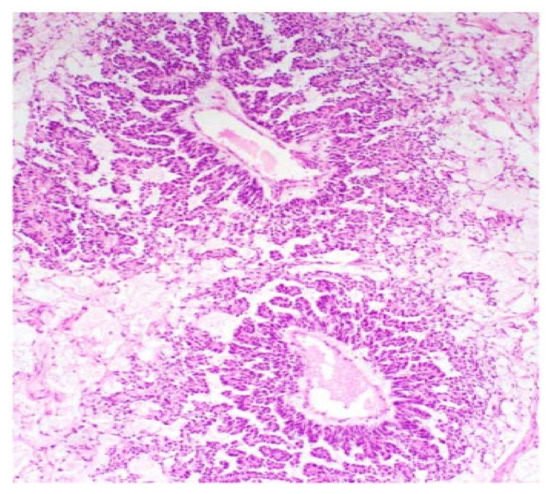
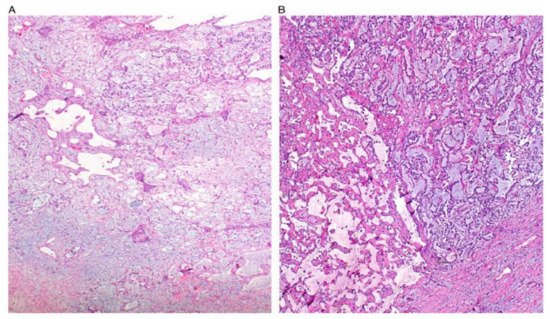
YST are the third most common GCT and mainly occur in women in their second and third decades of life [10,21][8][14]. YST is extremely rare in menopausal women but case reports of this are documented in the literature, with ages ranging between 50 and 86 years, and poorer outcomes have been identified for this demographic [22][15]. Patients will present with abdominal pain, a palpable abdominal mass, or an acute abdomen secondary to ovarian torsion [10][8]. Unlike dysgerminoma, serum AFP and CA-125 will be elevated in most patients [23][16]. Grossly, YST are unilateral (mostly), large, and well-encapsulated tumours, which have a mixture of solid and cystic components and areas of haemorrhage or necrosis [6][7]. The cysts vary in size and will be scattered throughout to give a wet honeycomb appearance [6][7]. The structure of YST resemble the primitive yolk sac and they have a variety of histological appearances, which are summarised in Table 2 [6,10,24][7][8][17]. Some figures are also provided (Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5). On CT and MRI imaging, YST will appear as a solid cystic mass with areas of haemorrhage and capsular tears may also be present [6,25][7][18]. Immunohistochemical staining for AFP is characteristic of YST [22][15]. YST is a highly malignant tumour which invades local structures and metastasises rapidly to intra-abdominal structures and retroperitoneal lymph nodes [10,26,27][8][19][20]. However, the combination of surgery with chemotherapy has now led to an overall cure rate of 80% [7,10,28][8][13][21].Figure 1. Reticular and microcystic (reticulocystic) patterns (A). Characteristic meshwork of spaces merging with cysts of varying sizes and shapes (B).
Figure 2.
Festoon pattern with arching drape-like configurations.
Figure 3. Solid pattern. Large focally coalescent aggregates are seen (A). Note typical abundant pale to clear cytoplasm and nuclei that are smaller than those of dysgerminoma (B).
Figure 4.
Papillary pattern. Occasionally the characteristic papillae radiated, like the spokes of a wheel, from a large dilated vessel.
Figure 5.
Myxoid appearance. Myxoid matrix occupying much of the stroma (
A
) and similar material within cyst lumens (
B
).
2.3. Treatment of GCT
Most GCT will be diagnosed at early stages and FSS is the main treatment option, especially as the patients affected are children or young women [13,29][22][23]. This includes unilateral salpingo-oophorectomy (USO) with the unaffected ovary and uterus left in place—biopsy of the second ovary is only performed if there is obvious abnormality to reduce the risk of adhesions or ovarian failure [6,10][7][8]. FSS requires further prospective evaluation, but multiple case studies have demonstrated successful outcomes with USO when disease is limited to one ovary [30,31][24][25]. Furthermore, peritoneal surfaces, the omentum, and lymph nodes (retroperitoneal and ipsilateral pelvic) are thoroughly examined and resected or biopsied if any abnormalities are present [6,32][7][26]. Surgical staging also includes peritoneal washings and ascites sampling if present [10][8]. If advanced disease is present, debulking surgery is performed instead to try and remove as much cancer as possible and second-look surgery may be an option if the cancer is not resected completely [10][8].
Depending on the histology, staging and molecular features of the GCT, surgery will be followed by active surveillance or adjuvant chemotherapy, which has revolutionised GCT treatment in the last 40 years [5,6][7][27]. Currently, dysgerminomas confined to the ovary and grade I immature teratoma are managed with surveillance post-operatively [5][27]. Otherwise, platinum-based chemotherapy is used, and the standard regime consists of bleomycin, etoposide and cisplatin (BEP) for four to six cycles [6,33][7][28]. Survival rates with BEP range between 82% to 100% in early-stage disease and 75% in advanced-stage disease [34][29].
Recurrences usually occur within two years of initial diagnosis and typically relapse in the peritoneal cavity and retroperitoneal lymph nodes. The response rate to salvage chemotherapy in patients with GCT is approximately 50%, and recommended regimens include vinblastine, ifosfamide, and cisplatin; etoposide, ifosfamide, and cisplatin; and paclitaxel, ifosfamide, and cisplatin. Secondary cytoreductive surgery could be performed in selected patients with recurrent disease [39][30].
3. Ovarian SCST
3.1. Ovarian GrCT
GrCT account for up to 5% of overall ovarian cancers. There is an incidence of 0.61 cases per 100,000 women per year, accounting for around 2–5% of all ovarian cancers [43][31]. Adult GrCT are the most common form, comprising around 95% of GrCT. The majority of these present at an early stage, in an indolent fashion. The most common presenting complaints with SCST are the presence of an adnexal mass, abdominal pain and distention. Some of these tumours are termed “functioning” and can release hormones, including androgens and oestrogens. As such, patients may have evidence of oestrogen excess, including abnormal vaginal bleeding, precocious puberty, or androgen excess, including hirsutism [44,45][32][33]. The presence of some tumour markers can be of diagnostic benefit. Inhibin A and B, oestradiol and MIH (Müllerian inhibiting substance) are hormones secreted by GrCT. MIH levels can also be elevated, and the combination of inhibins and MIH is useful for monitoring disease status. Adult GrCT have been found to have an association with a somatic c.402C > G missense point mutation in the FOXL2 gene. This is pathognomonic for adult GrCT and may have a causative role [48][34]. In addition, genetic studies can be used to differentiate between early stage and advanced stage adult GrCT. Due to oestrogen production by GrCT, there can be an associated finding of endometrial hyperplasia, and endometrial carcinoma can be seen in up to 10% of patients with GrCT. These are often at an early stage, and thus carry a good prognosis [49][35]. Surgical management is used to treat GrCT. Patients who present in their reproductive years are managed with a USO. For patients who are postmenopausal or have no desire of childbearing, they are treated with a total abdominal hysterectomy and bilateral salpingo-oophorectomy (BSO) [45][33]. For patients with stage IA GrCT, surgery alone provides an excellent prognosis, with no adjuvant therapies used. Around 97% of GrCT are unilateral [50][36]. For stage IB, i.e., involving both ovaries, a total abdominal hysterectomy with BSO is used. FSS can be attempted in selected cases. For stage IC, surgery alongside adjuvant platinum-based chemotherapy is used. For advanced or metastatic disease, cytoreductive surgery is the most effective treatment. Additional chemotherapy courses can be used in these patients, as the prognosis is poor, with a high recurrence rate [2]. Hormone therapy has been shown to have a role in GrCT which express steroid hormone receptors. No corroboration between hormonal therapy and hormone receptor expression has been established. Aromatase inhibitors have been found to be the most responsive hormone therapy, though data is limited [51][37]. GrCT have a good prognosis as they are usually diagnosed at an early stage. There are numerous significant prognostic factors related to GrCT, including the stage of the tumour, size of the tumour, whether or not it has ruptured, the mitotic index, and the presence of residual disease following surgical resection and grading. The most accurate prognostic factor in GrCT is the staging of the tumour at the time of diagnosis [52][38]. An early stage is also associated with reduced recurrence rates [45][33].3.2. Sertoli-Leydig Cell Tumours
SLCT are rare; they account for less than 0.5% of ovarian tumours. The majority occur in women aged between 20 and 40. Their specific incidence is not easily determined. They often present with a unilateral mass. The tumours are usually hormone-secreting; the majority secrete testosterone but some may produce oestradiol. Up to 85% of patients experience virilisation. Patients may have pure Sertoli cell tumours which can secrete renin, which can cause hypertension [40][39]. SLCT are formed of varying proportions of Sertoli cells and Leydig cells, and may have other heterogeneous elements [56][40]. These cells are normally found around the seminiferous tubules of the testicles, and are involved in androgen production. SLCT are classified into well-differentiated, moderately differentiated and poorly differentiated tumours. Both moderately- and poorly-differentiated SLCT express at least mutation in the DICER1 gene, whilst well-differentiated SLCT can be DICER1-independent. The association between the DICER1 mutation and SLCT has been shown to be around 88% [57][41]. The FOXL2 missense mutation, which is associated with GrCT can also be found in SLCT, but the sensitivity is only around 50%. As such, FOXL2 is generally used with serum inhibins to distinguish SCST from non-SCST, rather than to compare different SLCT [58][42]. For young patients who have stage IA SLCT and are of reproductive age, FSS is offered. Those with poorly differentiated malignancy are offered adjuvant chemotherapy. Patients who have no desire of childbearing are offered a total abdominal hysterectomy and BSO. For SLCT staging greater than IA, surgery and adjuvant chemotherapy are offered irrespective of tumour differentiation. The chemotherapy regimens are platinum-based [2]. SLCT carry a good prognosis since most patients at time of diagnosis have a disease stage of I; nevertheless, advanced stages carry an extremely poor prognosis [59][43].4. Small Cell Ovarian Carcinomas
Small cell carcinomas of the ovary hypercalcaemic type (SCCOHT) typically occur in adolescents and young women with a peak incidence in the third decade of life. Serum calcium levels may serve as a marker for treatment response and recurrences. The pathophysiology of hypercalcemia is unclear, it has been postulated that the parathyroid hormone or parathyroid hormone-related protein (PTHrP) appear to be crucial [40][39]. The prognosis of SCCOHT is very poor and the risk of extra-ovarian spread high [2]. A germ cell origin for SCCOHT has been suggested. However, more recently, SCCOHT have been sequenced and confirmed to be a malignant rhabdoid tumour by virtue of consistent deleterious mutations in SMARCA4, a chromatin-remodelling gene encoding protein BRG1. These tumours resemble high grade neuroendocrine histological features; nevertheless, they are now recognised to be a distinct clinical and pathological entity. They are predominantly characterised by variable numbers of larger cells with a luteinised or rhabdoid appearance [40][39]. There is currently no consensus as far as the treatment of the SCCOHT is concerned. A combination of treatment modalities, consisting of debulking surgery, followed by chemotherapy and possibly radiotherapy is recommended. FSS is reasonable, as the disease is mostly unilateral [40][39]. A combination of a cisplatin and etoposide-based therapy is generally considered most appropriate. SCCOHT are particularly chemosensitive at the outset but the relapse is usually rapid. Small cell carcinomas of the ovary, pulmonary type (SCCOPT) affect older, peri- or postmenopausal patients. In contrast with SCCOHT, they are not correlated with hypercalcemia. SCCOPT are predominantly unilateral and have dismal prognosis even when diagnosed early. Histopathologically, they have solid growth of small cells arranged in sheets and closely packed nests. The tumour cells are pleomorphic, round to spindle-shaped, with scanty cytoplasm and hyperchromatic and elongated nuclei. Pre-existing endometrioid carcinomas and Brenner tumours are predisposition factors for the development of the SCCOPT. Neuroendocrine markers (chromogranin, synaptophysin and NSE) are diffusely positive by immunohistochemistry [40][39]. Conventional surgical treatment includes radical surgery (hysterectomy and BSO), followed by adjuvant platinum-etoposide chemotherapy. The evidence for the potential role of the immunotherapy is extrapolated from the small-cell lung cancers. It seems that there is a promising activity of the anti-PD1 antibodies in this context [2].References
- Bennetsen, A.K.K.; Baandrup, L.; Aalborg, G.L.; Kjaer, S.K. Non-epithelial ovarian cancer in Denmark—Incidence and survival over nearly 40 years. Gynecol. Oncol. 2020, 157, 693–699.
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. ESMO Guidelines Committee. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv1–iv18.
- Boussios, S.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Tsiouris, A.K.; Chatziantoniou, A.A.; Kanellos, F.S.; Tatsi, K. Ovarian carcinosarcoma: Current developments and future perspectives. Crit. Rev. Oncol. Hematol. 2019, 134, 46–55.
- Samartzis, E.P.; Labidi-Galy, S.I.; Moschetta, M.; Uccello, M.; Kalaitzopoulos, D.R.; Perez-Fidalgo, J.A.; Boussios, S. Endometriosis-associated ovarian carcinomas: Insights into pathogenesis, diagnostics, and therapeutic targets-a narrative review. Ann. Transl. Med. 2020, 8, 1712.
- Matz, M.; Coleman, M.P.; Sant, M.; Chirlaque, M.D.; Visser, O.; Gore, M.; Allemani, C.; the CONCORD Working Group. The histology of ovarian cancer: Worldwide distribution and implications for international survival comparisons (CONCORD-2). Gynecol. Oncol. 2017, 144, 405–413.
- Vicus, D.; Beiner, M.E.; Klachook, S.; Le, L.W.; Laframboise, S.; Mackay, H. Pure dysgerminoma of the ovary 35 years on: A single institutional experience. Gynecol. Oncol. 2010, 117, 23–26.
- Shaaban, A.M.; Rezvani, M.; Elsayes, K.M.; Baskin, H., Jr.; Mourad, A.; Foster, B.R.; Jarboe, E.A.; Menias, C.O. Ovarian malignant germ cell tumors: Cellular classification and clinical and imaging features. Radiographics 2014, 34, 777–801.
- Kaur, B. Pathology of malignant ovarian germ cell tumours. Diagn. Histopathol. 2020, 26, 289–297.
- Kim, S.H.; Kang, S.B. Ovarian dysgerminoma: Color Doppler ultrasonographic findings and comparison with CT and MR imaging findings. J. Ultrasound Med. 1995, 14, 843–848.
- Guerriero, S.; Testa, A.C.; Timmerman, D.; Van Holsbeke, C.; Ajossa, S.; Fischerova, D.; Franchi, D.; Leone, F.P.; Domali, E.; Alcazar, J.L.; et al. Imaging of gynecological disease (6): Clinical and ultrasound characteristics of ovarian dysgerminoma. Ultrasound Obstet. Gynecol. 2011, 37, 596–602.
- Tîrnovanu, M.C.; Florea, I.D.; Tănase, A.; Toma, B.F.; Cojocaru, E.; Ungureanu, C.; Lozneanu, L. Un-common Metastasis of Ovarian Dysgerminoma: A Case Report and Review of the Literature. Medicina 2021, 57, 534.
- Chan, J.K.; Tewari, K.S.; Waller, S.; Cheung, M.K.; Shin, J.Y.; Osann, K.; Kapp, D.S. The influence of conservative surgical practices for malignant ovarian germ cell tumors. J. Surg. Oncol. 2008, 98, 111–116.
- Euscher, E.D. Germ Cell Tumors of the Female Genital Tract. Surg. Pathol. Clin. 2019, 12, 621–649.
- Kurman, R.J.; Norris, H.J. Endodermal sinus tumor of the ovary: A clinical and pathologic analysis of 71 cases. Cancer 1976, 38, 2404–2419.
- Boussios, S.; Attygalle, A.; Hazell, S.; Moschetta, M.; McLachlan, J.; Okines, A.; Banerjee, S. Malignant Ovarian Germ Cell Tumors in Postmenopausal Patients: The Royal Marsden Experience and Literature Review. Anticancer Res. 2015, 35, 6713–6722.
- Chao, W.T.; Liu, C.H.; Lai, C.R.; Chen, Y.J.; Chuang, C.M.; Wang, P.H. Alpha-fetoprotein-producing ovarian clear cell adenocarcinoma with fetal gut differentiation: A rare case report and literature review. J. Ovarian Res. 2018, 11, 52.
- Rakusić, Z.; Krpan, A.M.; Mareković, Z.; Juretić, A.; Gasparov, S. Retroperitoneal and metachronous testicular germ cell tumors with different histology and teratoma growing syndrome—A case report. Coll. Antropol. 2011, 35, 937–940.
- Li, Y.; Zheng, Y.; Lin, J.; Xu, G.; Cai, A.; Chen, R.; Wu, M. Radiological-pathological correlation of yolk sac tumor in 20 patients. Acta Radiol. 2016, 57, 98–106.
- Umezu, T.; Kajiyama, H.; Terauchi, M.; Shibata, K.; Ino, K.; Nawa, A.; Kikkawa, F. Long-term outcome and prognostic factors for yolk sac tumor of the ovary. Nagoya J. Med. Sci. 2008, 70, 29–34.
- McBee, W.C., Jr.; Brainard, J.; Sawady, J.; Rose, P.G. Yolk sac tumor of the ovary associated with endometrioid carcinoma with metastasis to the vagina: A case report. Gynecol. Oncol. 2007, 105, 244–247.
- Chen, L.H.; Yip, K.C.; Wu, H.J.; Yong, S.B. Yolk Sac Tumor in an Eight-Year-Old Girl: A Case Report and Literature Review. Front. Pediatr. 2019, 7, 169.
- Dellino, M.; Silvestris, E.; Loizzi, V.; Paradiso, A.; Loiacono, R.; Minoia, C.; Daniele, A.; Cormio, G. Germinal ovarian tumors in reproductive age women: Fertility-sparing and outcome. Medicine 2020, 99, e22146.
- Turkmen, O.; Karalok, A.; Basaran, D.; Kimyon, G.C.; Tasci, T.; Ureyen, I.; Tulunay, G.; Turan, T. Fertility-Sparing Surgery Should Be the Standard Treatment in Patients with Malignant Ovarian Germ Cell Tumors. J. Adolesc. Young Adult Oncol. 2017, 6, 270–276.
- Johansen, G.; Dahm-Kähler, P.; Staf, C.; Flöter Rådestad, A.; Rodriguez-Wallberg, K.A. Fertility-sparing surgery for treatment of non-epithelial ovarian cancer: Oncological and reproductive outcomes in a prospective nationwide population-based cohort study. Gynecol. Oncol. 2019, 155, 287–293.
- Park, J.Y.; Kim, D.Y.; Suh, D.S.; Kim, J.H.; Kim, Y.M.; Kim, Y.T.; Nam, J.H. Outcomes of Surgery Alone and Surveillance Strategy in Young Women with Stage I Malignant Ovarian Germ Cell Tumors. Int. J. Gynecol. Cancer 2016, 26, 859–864.
- Pectasides, D.; Pectasides, E.; Kassanos, D. Germ cell tumors of the ovary. Cancer Treat. Rev. 2008, 34, 427–441.
- Veneris, J.T.; Mahajan, P.; Frazier, A.L. Contemporary management of ovarian germ cell tumors and remaining controversies. Gynecol. Oncol. 2020, 158, 467–475.
- Kang, H.; Kim, T.J.; Kim, W.Y.; Choi, C.H.; Lee, J.W.; Kim, B.G.; Bae, D.S. Outcome and reproductive function after cumulative high-dose combination chemotherapy with bleomycin, etoposide and cisplatin (BEP) for patients with ovarian endodermal sinus tumor. Gynecol. Oncol. 2008, 111, 106–110.
- Smith, H.O.; Berwick, M.; Verschraegen, C.F.; Wiggins, C.; Lansing, L.; Muller, C.Y.; Qualls, C.R. Incidence and survival rates for female malignant germ cell tumors. Obstet. Gynecol. 2006, 107, 1075–1085.
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41.
- Van Meurs, H.S.; Bleeker, M.C.; van der Velden, J.; Overbeek, L.I.; Kenter, G.G.; Buist, M.R. The incidence of endometrial hyperplasia and cancer in 1031 patients with a granulosa cell tumor of the ovary: Long-term follow-up in a population-based cohort study. Int. J. Gynecol. Cancer 2013, 23, 1417–1422.
- Outwater, E.K.; Wagner, B.J.; Mannion, C.; McLarney, J.K.; Kim, B. Sex cord-stromal and steroid cell tumors of the ovary. Radiographics 1998, 18, 1523–1546.
- Khosla, D.; Dimri, K.; Pandey, A.K.; Mahajan, R.; Trehan, R. Ovarian granulosa cell tumor: Clinical features, treatment, outcome, and prognostic factors. N. Am. J. Med. Sci. 2014, 6, 133–138.
- Shah, S.P.; Köbel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009, 360, 2719–2729.
- Schumer, S.T.; Cannistra, S.A. Granulosa cell tumor of the ovary. J. Clin. Oncol. 2003, 21, 1180–1189.
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial Ovarian Cancer: Elucidating Uncommon Gynaecological Malignancies. Anticancer Res. 2016, 36, 5031–5042.
- Van Meurs, H.S.; van Lonkhuijzen, L.R.; Limpens, J.; van der Velden, J.; Buist, M.R. Hormone therapy in ovarian granulosa cell tumors: A systematic review. Gynecol. Oncol. 2014, 134, 196–205.
- Zhang, M.; Cheung, M.K.; Shin, J.Y.; Kapp, D.S.; Husain, A.; Teng, N.N.; Berek, J.S.; Osann, K.; Chan, J.K. Prognostic factors responsible for survival in sex cord stromal tumors of the ovary-an analysis of 376 women. Gynecol. Oncol. 2007, 104, 396–400.
- Boussios, S.; Moschetta, M.; Zarkavelis, G.; Papadaki, A.; Kefas, A.; Tatsi, K. Ovarian sex-cord stromal tumours and small cell tumours: Pathological, genetic and management aspects. Crit. Rev. Oncol. Hematol. 2017, 120, 43–51.
- Roth, L.M. Recent advances in the pathology and classification of ovarian sex cord-stromal tumors. Int. J. Gynecol. Pathol. 2006, 25, 199–215.
- De Kock, L.; Terzic, T.; McCluggage, W.G.; Stewart, C.J.R.; Shaw, P.; Foulkes, W.D.; Clarke, B.A. DICER1 Mutations Are Consistently Present in Moderately and Poorly Differentiated Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2017, 41, 1178–1187.
- Al-Agha, O.M.; Huwait, H.F.; Chow, C.; Yang, W.; Senz, J.; Kalloger, S.E.; Huntsman, D.G.; Young, R.H.; Gilks, C.B. FOXL2 is a sensitive and specific marker for sex cord-stromal tumors of the ovary. Am. J. Surg. Pathol. 2011, 35, 484–494.
- Colombo, N.; Parma, G.; Zanagnolo, V.; Insinga, A. Management of ovarian stromal cell tumors. J. Clin. Oncol. 2007, 25, 2944–2951.
More