Cancer of unknown primary (CUP) encloses a group of heterogeneous tumours, the primary sites for which cannot be identified at the time of diagnosis, despite extensive investigations. CUP has always posed major challenges both in its diagnosis and management, leading to the hypothesis that it is rather a distinct entity with specific genetic and phenotypic aberrations, considering the regression or dormancy of the primary tumour; the development of early, uncommon systemic metastases; and the resistance to therapy. Patients with CUP account for 1–3% of all human malignancies and can be categorised into two prognostic subsets according to their clinicopathologic characteristics at presentation. The diagnosis of CUP mainly depends on the standard evaluation comprising a thorough medical history; complete physical examination; histopathologic morphology and algorithmic immunohistochemistry assessment; and CT scan of the chest, abdomen, and pelvis.
- cancer of unknown primary (CUP)
- biology
- molecular profiling
- classification
- diagnosis
- treatment
1. Introduction
2. Epidemiology of CUP
From the epidemiological perspective, around 8600 new CUPs are diagnosed each year in the United Kingdom, making this the fifteenth most prevalent cancer [8]. In 2012, Denmark had around 338 new cases per 100,000 people, compared to 284 in Germany, 296 in Canada, and 318 in the United States [9]. Since the early 1980s, CUP incidence rates in the United States have been falling at a pace of 3.6% per year over the previous two decades. Since 1973, the rate of non-microscopically confirmed CUP has decreased by 2.6% every year [10]. CUP incidence rates in Scotland climbed from 7 to 8 per 100,000 in the 1960s to a peak of 14–18 per 100,000 in the early to mid-1990s, before falling precipitously to 8 per 100,000 in 2009 [11]. Between 1999 and 2017, the incidence of CUP decreased in Korea. CUP incidence probably decreased due to improved diagnostics, which have led to better identification of the primary culprit. Genomic profiling testing may help in identifying molecular signatures in CUP patients and enable targeted treatment [12]. Patients aged 80 and up had the highest risk of occurrence. The survival rate climbed from 14.2% in 1999–2002 to 27.3% in 2013–2017 [13]. In Sweden, it is estimated that the age-standardised incidence increased from 10 per 100,000 in the early 1960s to 16 per 100,000 around the year 2000 for both males and females [14].3. Risk Factors of CUP
Any degree of smoking raises the chance of CUP with respiratory system metastases to 4.9%. In a study, smoking was strongly linked to an increased risk of CUP, with a relative risk of 3.66% for ongoing, heavy smokers (>25 cigarettes/day) compared to never smokers (standardised for other risk factors), and a relative risk of 5.12% for patients with CUP who died within less 12 months since the diagnosis [15]. Other risk factors include alcohol intake, body mass index (BMI), waist circumference, diabetes, and a poor educational level or socioeconomic position [16]. The pathophysiology of familial CUP is characterised by the existence of a genetic vulnerability that puts family members of a CUP patient at a higher risk of CUP and other tumours [17]. Relatives of CUP patients are more likely to develop CUP as well as other malignant cancers, especially of the lung, pancreas, and colon [18].4. Biology of CUP
Generally, the ongoing improved knowledge of the biology of several cancers has enabled a more-accurate classification, diagnosis, and prognosis, as well as providing guidance in the tailoring of specific therapies. However, in CUP, the data are not very mature. The early dissemination of tumour cells implies subsequent independent progression of the primary tumour and metastases, under the selection pressure of the immune system. The clinical observation of the high systemic relapse rate in CUP patients with localised disease treated with curative intention with surgery and/or radiotherapy supports that model. Still, molecular platforms represent a key component of the diagnostic work-up and clinical management [12].5. Classification of CUP
The minority of patients with CUP (15–20%) present with clinical and pathological features that can be attributed to a primary culprit (Table 1). The favourable risk cancer subgroup comprises peritoneal adenocarcinomatosis of a serous papillary subtype, isolated axillary nodal metastases in females, squamous cell carcinoma involving nonsupraclavicular cervical lymph nodes, single metastatic deposit from unknown primary, neuroendocrine carcinomas of unknown primary, and men with blastic bone metastases and elevated prostate-specific antigen (PSA). The treatment of these patients is compatible with the corresponding primary guidelines for metastatic disease. Currently, new favourable subsets of CUP have emerged, including colorectal, lung, and renal CUP, which underly specific treatments [47][19]. These patients generally harbour a chemosensitive disease and, as such, longer life expectancy.|
Favourable Subsets |
Unfavourable Subsets |
|
|---|---|---|
|
1 |
Poorly differentiated carcinoma with midline distribution (extragonadal germ cell syndrome) |
Adenocarcinoma metastatic to the liver or other organs |
|
2 |
Women with papillary adenocarcinoma of the peritoneal cavity |
Non-papillary malignant ascites (adenocarcinoma) |
|
3 |
Women with adenocarcinoma involving only axillary lymph nodes |
Multiple cerebral metastases (adeno or squamous carcinoma) |
|
4 |
Squamous cell carcinoma involving cervical lymph nodes |
Multiple lung/pleural metastases (adenocarcinoma) |
|
5 |
Isolated inguinal adenopathy (squamous carcinoma) |
Multiple metastatic bone disease (adenocarcinoma) |
|
6 |
Poorly differentiated neuroendocrine carcinomas |
Squamous abdominopelvic CUP |
|
7 |
Men with blastic bone metastases and elevated PSA (adenocarcinoma) |
|
|
8 |
Patients with a single, small, potentially resectable tumour |
|
|
9 |
CUP patients with a single small metastasis |
|
|
10 |
Merkel cell adenopathy of unknown origin |
6. Diagnostic Workup
The diagnosis of CUP is established when a metastatic cancer is histologically confirmed in the absence of identifiable primary tumour site, despite the extensive diagnostic evaluation. Recent research has focused on using genomics and transcriptomics to identify the origin of the primary tumour, but it is still not always performed, especially in low-resource environments [49][21]. The development of tissue of origin classifiers for the analysis and diagnostics of CUP using a whole genome sequencing dataset of both primary and metastatic tumours is still an effort in progress [4].6.1. Pathology and Immunohistochemistry
From the histological perspective, CUP is defined as well- or moderately differentiated adenocarcinomas, accounting for 50–70% of all cases, with poorly differentiated carcinomas and adenocarcinomas making up another 20–30%, and the remaining being squamous-cell carcinomas (5–8%) and undifferentiated malignant neoplasms (2–3%) (Figure 1) with inability of light microscopy to distinguish among carcinomas, lymphomas, melanomas, and sarcomas [47,50,51,52][19][22][23][24]. The diagnoses of neuroendocrine tumours, melanomas, and sarcomas can be based on immunoperoxidase staining.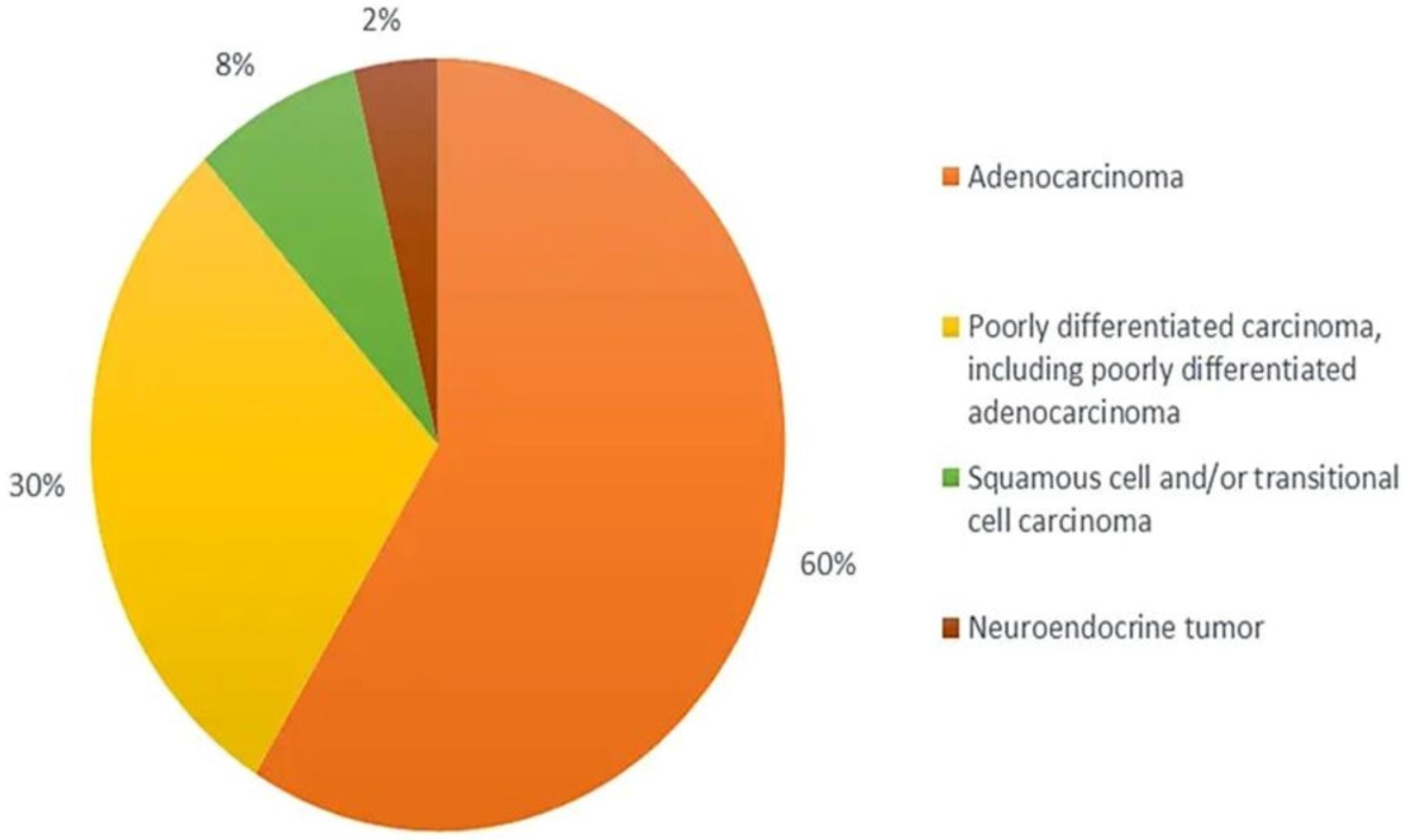
6.2. Diagnostic Radiology
Image-assisted technologies has revolutionised the diagnosis of CUP. CT and conventional MRI have both been used to locate lesions, considering the clinical manifestation of CUP. The diagnostic accuracy of CT scans is around 55% (36–74%), mainly in pancreatic, colorectal, and lung cancer, while MRI has a sensitivity of 70% in detecting primary breast cancers [56][25]. However, the diagnosis can be challenging if the primary tumour is small in size or has regressed, hindering the diagnosis. In particular, these cases may be successfully facilitated with the 2-[18F] fluoro-2-deoxy-d-glucose (FDG) PET/CT, but still the detection rate is around 40% [57][26]. The most frequent primary sites identified by PET are lung (33%) and head and neck (27%), followed by pancreas, breast, and colon (4–5%). Finally, 68Ga-DOTA-NOC receptor PET/CT is recommended for the identification of primary neuroendocrine tumours, along with their metastases [58][27]. Mammography is recommended for female patients with metastatic adenocarcinomas involving axillary lymph nodes. In patients with mammographically occult breast cancer, breast MRI may be considered.6.3. Endoscopy
Endoscopy should be directed towards investigating specific symptoms and signs or when specific histopathological findings are available. Fiberoptic bronchoscopy is reasonable for patients with respiratory symptoms and/or expression of CK7 and TTF1, whereas colonoscopy should be requested for those with abdominal symptoms or occult blood in the stool and/or expression of CK7, CK20, and CDX2. The sensitivity and specificity of the endoscopies are generally low.6.4. Serum Tumour Markers
In almost 70% of CUP patients, more than one marker can be concomitantly elevated in a non-specific way. Routine request of cancer antigen 125 (CA 125), cancer antigen 15.3 (CA 15-3), carbohydrate antigen 19-9 (CA 19-9), and carcinoembryonic antigen (CEA) is not recommended due to lack of prognostic and/or predictive value [59][28]. However, there are some clinical scenarios in which serum tumour markers may have some diagnostic value. Indeed, serum PSA should be evaluated in men with osteoblastic bone metastases, CA 125 in women with primary serous papillary peritoneal adenocarcinoma, and CA 15-3 in females with isolated axillary adenocarcinoma. Finally, a high level of thyroglobulin in patients with CUP and bone metastasis may indicate occult thyroid cancer [60][29].6.5. Liquid Biopsy
Improvements in nucleic acid sequencing technologies have enabled the detection of low quantities of tumour genetic material within the blood and show the potential to be both sensitive and specific to an individual’s tumour. These blood-based biomarkers include cfDNA, tumour microRNAs (miRNAs), and platelet-derived tumour mRNA, as well as analysis of DNA, RNAs, and protein expression from individual circulating tumour cells. Within this context, the use of liquid biopsies reduces the need for intrusive diagnostic biopsies and provides enough material to perform the diagnostic procedures. For instance, even though the presence of aberrant hypermethylation of tumour-suppressor genes in serum DNA has been detectable before the millennium, more sensitive and quantitative techniques for analysis of DNA methylation are required to expedite its incorporation in the clinical setting [61][30]. In diffuse large-B-cell lymphoma, detection of aberrant DAPK1 methylation in cfDNA at the time of diagnosis is a positive prognostic biomarker, whilst in hepatocellular carcinoma, methylation of VIM is an early detection biomarker [62,63][31][32]. Overall, there is evidence that the tissue of origin can be determined using cfDNA [64][33].6.6. Molecular Profiling for the Tissue of Origin
Molecular profiling technologies including microarray-based gene expression profiling, reverse transcriptase polymerase chain reaction, RNA sequencing, somatic gene mutation profiling with NGS, and DNA methylation profiling were used to define the primary culprit among patients with CUP. However, the implementation of tissue-of-origin classifiers in CUP is limited due to the absence of primary tumour. Some studies were conducted to validate predictions of the primary origin, on the basis of autopsy data, latent primary emergence, or IHC. Gene expression profiling has been directly compared to IHC, within known metastatic tumour types. Accuracy of gene expression profiling was 89%, compared with 83% for IHC when only one round of IHC determined the diagnosis, whereas in poorly differentiated cancers, such as CUP, the percentages were 83% and 67%, respectively [65][34]. Nevertheless, only a limited number of studies have investigated the clinical outcomes of CUP patients, treated on the basis of gene expression predictions [66][35]. Predicting the tissue-of-origin via molecular profiling is a debated topic within CUP, given that it is difficult to be molecularly classified in the absence of histological definition. However, the incorporation of molecular classifiers to the standard diagnostic workup may potentially identify atypical presentations of patients for whom site-specific therapies would be effective [67][36]. PlexinB2 (PlxnB2) is a semaphorin receptor implicated in the regulation of cancer cell proliferation, invasiveness, and metastatic spreading [68,69,70,71][37][38][39][40]. The G842C-PlxnB2 variant has been investigated in an effort to establish a proof of principle about the relevance of axon guidance genes in CUP. This mutation affected the conserved fold of an IPT domain, a moiety also found in Met and Ron oncogenic receptors [72,73][41][42]. Notably, the large intracellular portion of the plexins does not contain a kinase domain or other classical signalling domains; nevertheless, it regulates the activity of monomeric GTPases, especially R-Ras, Rap-1, and RhoA. Moreover, plexins have been shown to couple with transmembrane tyrosine kinases such as ErbB2 and Met, triggering alternative noncanonical signalling cascades, especially in cancer cells [74][43].7. Treatment of CUP
Traditionally, CUP patients who are classified into one of the favourable subsets are treated according to their corresponding primary guidelines for metastatic disease. CUP patients with poorly differentiated carcinoma with midline distribution (extragonadal germ cell syndrome) should be managed like poor prognosis germ cell tumours with platinum-based combination chemotherapy. More than 50% response has been reported, with 15–25% complete responders and 10–15% long-term disease-free survivors. Women with papillary adenocarcinoma of the peritoneal cavity are optimally treated as FIGO stage III ovarian cancer. The recommended strategy includes aggressive surgical cytoreduction, followed by platinum-based postoperative chemotherapy. The median response rate is 80%, whilst 30–40% of the patients are complete responders. Similarly to FIGO stage III ovarian cancer patients, the median survival is 36 months [76,77][44][45]. For the subgroup of women with adenocarcinoma involving only axillary lymph nodes, locoregional treatment with or without systemic therapy is suggested. The management is compatible with stage II/III breast cancer, resulting in 5- and 10-year overall survival rates of 75 and 60%, respectively. The patients with squamous cell carcinoma involving cervical lymph nodes are treated with locoregional management, according to the guidelines for locally advanced head and neck cancer. The 5-year survival rates range from 35 to 50% with documented long-term disease-free survivors. Surgery alone is inferior and only recommended in selected patients, particularly those with pN1 neck disease with no extracapsular extension. Radiotherapy to the ipsilateral cervical nodes alone is still inferior to extensive irradiation to both sides of the neck and the mucosa in the entire pharyngeal axis and larynx. Whether such intensive irradiation prolongs survival is still uncertain. Although the role of systemic chemotherapy remains undefined, concurrent chemoradiotherapy seems to be beneficial, particularly in patients with an N2 or N3 lymph node disease. The group of CUP patients with poorly differentiated neuroendocrine carcinomas should be treated with empirical platinum-based or platinum-taxane chemotherapy. The reported response rates are as high as 50–70% with 25% complete responders and 10–15% long-term survivors. Men with blastic bone metastases and elevated PSA are considered as having advanced/metastatic prostate cancer and treated accordingly. The appropriate approach of CUP patients with a single small metastasis is the local treatment with either resection and/or radiotherapy. A considerable number of these patients have a long disease-free survival [78][46]. Finally, the treatment of Merkel cell cancer (MCC) of unknown origin is largely multimodal in nature and includes surgery, radiotherapy, and chemotherapy. For primary MCC that is associated with clinically positive nodal disease or with positive sentinel node, complete dissection of the involved regional nodal basin is recommended [79][47]. MCC is radiosensitive, and as such, radiotherapy may be an alternative definitive treatment for medically ineligible surgical resection patients. In contrast, adjuvant chemotherapy has a limited role in MCC. The treatment of patients with unfavourable CUP subsets is usually empirical chemotherapy, consisting of either taxanes or platinum-based regimens, on the basis of randomised trials showing dismal survival improvements [80][48]. The biomarker-based approach has been considered using targeted-therapy; nevertheless, the available evidence is limited to anecdotal cases [81][49]. Site-specific therapy guided by molecular classifiers was evaluated in this context. A meta-analysis of two retrospective and two prospective trials evaluating site-specific treatments in CUP was performed [82][50]. A trend towards improved overall survival was noted with site-specific versus empiric treatment for CUP (hazard ratio (HR) = 0.73, 95% confidence interval (CI) 0.52–1.02). The results of this meta-analysis highlighted the significant heterogeneity between the prospective studies comparing molecularly tailored to empiric therapy for CUP. In the most up-to-date meta-analysis of five studies that included 1114 patients, site-specific therapy was not significantly associated with improved overall survival (HR 0.75, 95% CI 0.55–1.03, p = 0.069) compared with empiric therapy [83][51]. CUPISCO (NCT03498521) is an ongoing prospective, phase II, randomised study designed to elucidate the potential benefit of treatment following genomic profiling, as compared to standard chemotherapy of CUP patients [84][52]. The study includes an atezolizumab monotherapy arm for the tumour mutational burden-high patients and a combination chemotherapy/atezolizumab arm for patients with tumour mutational burden-low or unknown tumours. The study experienced severe issues in patients’ accrual, along with screen failures. Molecular analyses, such as the identification of currently non-targetable alterations that may affect disease dynamics or be correlated with resistance, should be performed. Within this context, immunotherapies have the potential to improve outcomes in this population, due to the PD-L1 expression and high tumour mutational burden in 22.5% and 11.8% of CUP patients, respectively [45][53]. Overall, the genomic mutation correlates of response and resistance to immune checkpoint inhibitors do not differ between CUP and tumours that are immune checkpoint inhibitor eligible [86][54]. Tumour mutational burden >10 mutations per megabase trended towards better outcomes in CUP patients treated with immune checkpoint inhibitors. Furthermore, MDM2 amplification, which is associated with lack of response to immune checkpoint inhibitors, has been detected in 2% of CUP patients [45][53].References
- Fizazi, K.; Greco, F.A.; Pavlidis, N.; Daugaard, G.; Oien, K.; Pentheroudakis, G.; ESMO Guidelines Committee. Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v133–v138.
- Rassy, E.; Pavlidis, N. The currently declining incidence of cancer of unknown primary. Cancer Epidemiol. 2019, 61, 139–141.
- van der Strate, I.; Kazemzadeh, F.; Nagtegaal, I.D.; Robbrecht, D.; van de Wouw, A.; Padilla, C.S.; Duijts, S.; Esteller, M.; Greco, F.A.; Pavlidis, N.; et al. International consensus on the initial diagnostic workup of cancer of unknown primary. Crit. Rev. Oncol. Hematol. 2023, 181, 103868.
- Pisacane, A.; Cascardi, E.; Berrino, E.; Polidori, A.; Sarotto, I.; Casorzo, L.; Panero, M.; Boccaccio, C.; Verginelli, F.; Benvenuti, S.; et al. Real-world histopathological approach to malignancy of undefined primary origin (MUO) to diagnose cancers of unknown primary (CUPs). Virchows Arch. 2022, 1–13.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Occult Primary (Cancer of Unknown Primary ). NCCN. Version 1.2022—2 September 2021. Available online: http://www.nccn.org/professionals/physician_gls/pdf/occult.pdf (accessed on 11 December 2022).
- Losa, F.; Soler, G.; Casado, A.; Estival, A.; Fernández, I.; Giménez, S.; Longo, F.; Pazo-Cid, R.; Salgado, J.; Seguí, M.Á. SEOM clinical guideline on unknown primary cancer (2017). Clin. Transl. Oncol. 2018, 20, 89–96.
- National Institute for Health and Care Excellence (NICE). Surveillance Report (Exceptional Review) 2017—Metastatic Malignant Disease of Unknown Primary Origin in Adults (2010) NICE Guideline CG104; National Institute for Health and Care Excellence (NICE): London, UK, 2017.
- Cancer Research UK. 2022. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/cancer-of-unknown-primary#heading-Zero (accessed on 1 February 2023).
- Dyrvig, A.K.; Yderstræde, K.B.; Gerke, O.; Jensen, P.B.; Hess, S.; Høilund-Carlsen, P.F.; Green, A. Cancer of unknown primary: Registered procedures compared with national integrated cancer pathway for illuminating external validity. Medicine 2017, 96, e6693.
- Mnatsakanyan, E.; Tung, W.C.; Caine, B.; Smith-Gagen, J. Cancer of unknown primary: Time trends in incidence, United States. Cancer Causes Control 2014, 25, 747–757.
- Brewster, D.H.; Lang, J.; Bhatti, L.A.; Thomson, C.S.; Oien, K.A. Descriptive epidemiology of cancer of unknown primary site in Scotland, 1961–2010. Cancer Epidemiol. 2014, 38, 227–234.
- Moran, S.; Martinez-Cardús, A.; Boussios, S.; Esteller, M. Precision medicine based on epigenomics: The paradigm of carcinoma of unknown primary. Nat. Rev. Clin. Oncol. 2017, 14, 682–694.
- Boo, Y.K.; Park, D.; Lim, J.; Lim, H.S.; Won, Y.J. Descriptive epidemiology of cancer of unknown primary in South Korea, 1999–2017. Cancer Epidemiol. 2021, 74, 102000.
- Randn, M.; Rutqvist, L.E.; Johansson, H. Cancer patients without a known primary: Incidence and survival trends in Sweden 1960–2007. Acta Oncol. 2009, 48, 915–920.
- Agudo, A.; Bonet, C.; Travier, N.; González, C.A.; Vineis, P.; Bueno-de-Mesquita, H.B.; Trichopoulos, D.; Boffetta, P.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Impact of cigarette smoking on cancer risk in the European prospective investigation into cancer and nutrition study. J. Clin. Oncol. 2012, 30, 4550–4557.
- Kaaks, R.; Sookthai, D.; Hemminki, K.; Krämer, A.; Boeing, H.; Wirfält, E.; Weiderpass, E.; Overvad, K.; Tjønneland, A.; Olsen, A.; et al. Risk factors for cancers of unknown primary site: Results from the prospective EPIC cohort. Int. J. Cancer 2014, 135, 2475–2481.
- Rassy, E.; Kattan, J.; Pavlidis, N. Familial cancer of unknown primary. Int. J. Clin. Oncol. 2019, 24, 1328–1331.
- Samadder, N.J.; Smith, K.R.; Hanson, H.; Pimentel, R.; Wong, J.; Boucher, K.; Akerley, W.; Gilcrease, G.; Ulrich, C.M.; Burt, R.W.; et al. Familial Risk in Patients With Carcinoma of Unknown Primary. JAMA Oncol. 2016, 2, 340–346.
- Rassy, E.; Parent, P.; Lefort, F.; Boussios, S.; Baciarello, G.; Pavlidis, N. New rising entities in cancer of unknown primary: Is there a real therapeutic benefit? Crit. Rev. Oncol. Hematol. 2020, 147, 102882.
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435.
- Nguyen, L.; Van Hoeck, A.; Cuppen, E. Machine learning-based tissue of origin classification for cancer of unknown primary diagnostics using genome-wide mutation features. Nat. Commun. 2022, 13, 4013.
- Boussios, S.; Rassy, E.; Samartzis, E.; Moschetta, M.; Sheriff, M.; Pérez-Fidalgo, J.A.; Pavlidis, N. Melanoma of unknown primary: New perspectives for an old story. Crit. Rev. Oncol. Hematol. 2021, 158, 103208.
- Rassy, E.; Boussios, S.; Chebly, A.; Farra, C.; Kattan, J.; Pavlidis, N. Comparative genomic characterization of melanoma of known and unknown primary. Clin. Transl. Oncol. 2021, 23, 2302–2308.
- Rassy, E.; Abou-Jaoude, R.; Boussios, S.; Assi, T.; Kattan, J.; Khaled, H.; Pavlidis, N. Sarcoma of unknown primary: Myth or reality? J. Egypt. Natl. Canc. Inst. 2022, 34, 27.
- Pavlidis, N.; Fizazi, K. Carcinoma of unknown primary (CUP). Crit. Rev. Oncol. Hematol. 2009, 69, 271–278.
- Kwee, T.C.; Kwee, R.M. Combined FDG-PET/CT for the detection of unknown primary tumors: Systematic review and meta-analysis. Eur. Radiol. 2009, 19, 731–744.
- Prasad, V.; Ambrosini, V.; Hommann, M.; Hoersch, D.; Fanti, S.; Baum, R.P. Detection of unknown primary neuroendocrine tumours (CUP-NET) using (68)Ga-DOTA-NOC receptor PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 67–77.
- Takamizawa, S.; Shimoi, T.; Yoshida, M.; Tokura, M.; Yazaki, S.; Mizoguchi, C.; Saito, A.; Kita, S.; Yamamoto, K.; Kojima, Y.; et al. Diagnostic value of tumor markers in identifying favorable or unfavorable subsets in patients with cancer of unknown primary: A retrospective study. BMC Cancer 2022, 22, 412.
- Oltmann, S.C.; Leverson, G.; Lin, S.H.; Schneider, D.F.; Chen, H.; Sippel, R.S. Markedly elevated thyroglobulin levels in the preoperative thyroidectomy patient correlates with metastatic burden. J. Surg. Res. 2014, 187, 1–5.
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70.
- Kristensen, L.S.; Hansen, J.W.; Kristensen, S.S.; Tholstrup, D.; Harsløf, L.B.; Pedersen, O.B.; De Nully Brown, P.; Grønbæk, K. Aberrant methylation of cell-free circulating DNA in plasma predicts poor outcome in diffuse large B cell lymphoma. Clin. Epigenetics 2016, 8, 95.
- Vaca-Paniagua, F.; Oliver, J.; Nogueira da Costa, A.; Merle, P.; McKay, J.; Herceg, Z.; Holmila, R. Targeted deep DNA methylation analysis of circulating cell-free DNA in plasma using massively parallel semiconductor sequencing. Epigenomics 2015, 7, 353–362.
- Lubotzky, A.; Zemmour, H.; Neiman, D.; Gotkine, M.; Loyfer, N.; Piyanzin, S.; Ochana, B.L.; Lehmann-Werman, R.; Cohen, D.; Moss, J.; et al. Liquid biopsy reveals collateral tissue damage in cancer. JCI Insight 2022, 7, e153559.
- Handorf, C.R.; Kulkarni, A.; Grenert, J.P.; Weiss, L.M.; Rogers, W.M.; Kim, O.S.; Monzon, F.A.; Halks-Miller, M.; Anderson, G.G.; Walker, M.G.; et al. A multicenter study directly comparing the diagnostic accuracy of gene expression profiling and immunohistochemistry for primary site identification in metastatic tumors. Am. J. Surg. Pathol. 2013, 37, 1067–1075.
- Conway, A.M.; Mitchell, C.; Kilgour, E.; Brady, G.; Dive, C.; Cook, N. Molecular characterisation and liquid biomarkers in Carcinoma of Unknown Primary (CUP): Taking the ‘U’ out of ‘CUP’. Br. J. Cancer 2019, 120, 141–153.
- Rassy, E.; Labaki, C.; Chebel, R.; Boussios, S.; Smith-Gagen, J.; Greco, F.A.; Pavlidis, N. Systematic review of the CUP trials characteristics and perspectives for next-generation studies. Cancer Treat. Rev. 2022, 107, 102407.
- Le, A.P.; Huang, Y.; Pingle, S.C.; Kesari, S.; Wang, H.; Yong, R.L.; Zou, H.; Friedel, R.H. Plexin-B2 promotes invasive growth of malignant glioma. Oncotarget 2015, 6, 7293–7304.
- Yu, W.; Goncalves, K.A.; Li, S.; Kishikawa, H.; Sun, G.; Yang, H.; Vanli, N.; Wu, Y.; Jiang, Y.; Hu, M.G.; et al. Plexin-B2 Mediates Physiologic and Pathologic Functions of Angiogenin. Cell 2017, 171, 849–864.
- Gurrapu, S.; Pupo, E.; Franzolin, G.; Lanzetti, L.; Tamagnone, L. Sema4C/PlexinB2 signaling controls breast cancer cell growth, hormonal dependence and tumorigenic potential. Cell Death Differ. 2018, 25, 1259–1275.
- Huang, Y.; Tejero, R.; Lee, V.K.; Brusco, C.; Hannah, T.; Bertucci, T.B.; Junqueira Alves, C.; Katsyv, I.; Kluge, M.; Foty, R.; et al. Plexin-B2 facilitates glioblastoma infiltration by modulating cell biomechanics. Commun. Biol. 2021, 4, 145.
- Ma, Q.; Zhang, K.; Yao, H.P.; Zhou, Y.Q.; Padhye, S.; Wang, M.H. Inhibition of MSP-RON signaling pathway in cancer cells by a novel soluble form of RON comprising the entire sema sequence. Int. J. Oncol. 2010, 36, 1551–1561.
- Navis, A.C.; van Lith, S.A.; van Duijnhoven, S.M.; de Pooter, M.; Yetkin-Arik, B.; Wesseling, P.; Hendriks, W.J.; Venselaar, H.; Timmer, M.; van Cleef, P.; et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. 2015, 130, 131–144.
- Cagnoni, G.; Tamagnone, L. Semaphorin receptors meet receptor tyrosine kinases on the way of tumor progression. Oncogene 2014, 33, 4795–4802.
- Pavlidis, N.; Rassy, E.; Vermorken, J.B.; Assi, T.; Kattan, J.; Boussios, S.; Smith-Gagen, J. The outcome of patients with serous papillary peritoneal cancer, fallopian tube cancer, and epithelial ovarian cancer by treatment eras: 27 years data from the SEER registry. Cancer Epidemiol. 2021, 75, 102045.
- Rassy, E.; Assi, T.; Boussios, S.; Kattan, J.; Smith-Gagen, J.; Pavlidis, N. Narrative review on serous primary peritoneal carcinoma of unknown primary site: Four questions to be answered. Ann. Transl. Med. 2020, 8, 1709.
- Pouyiourou, M.; Wohlfromm, T.; Kraft, B.; Hielscher, T.; Stichel, D.; von Deimling, A.; Delorme, S.; Endris, V.; Neumann, O.; Stenzinger, A.; et al. Local ablative treatment with surgery and/or radiotherapy in single-site and oligometastatic carcinoma of unknown primary. Eur. J. Cancer 2021, 157, 179–189.
- Deneve, J.L.; Messina, J.L.; Marzban, S.S.; Gonzalez, R.J.; Walls, B.M.; Fisher, K.J.; Chen, Y.A.; Cruse, C.W.; Sondak, V.K.; Zager, J.S. Merkel cell carcinoma of unknown primary origin. Ann. Surg. Oncol. 2012, 19, 2360–2366.
- Hainsworth, J.D.; Daugaard, G.; Lesimple, T.; Hübner, G.; Greco, F.A.; Stahl, M.J.; Büschenfelde, C.M.; Allouache, D.; Penel, N.; Knoblauch, P.; et al. Paclitaxel/carboplatin with or without belinostat as empiric first-line treatment for patients with carcinoma of unknown primary site: A randomized, phase 2 trial. Cancer 2015, 121, 1654–1661.
- El Rassy, E.; Pavlidis, N. The current evidence for a biomarker-based approach in cancer of unknown primary. Cancer Treat. Rev. 2018, 67, 21–28.
- Rassy, E.; Bakouny, Z.; Choueiri, T.K.; Van Allen, E.M.; Fizazi, K.; Greco, F.A.; Pavlidis, N. The role of site-specific therapy for cancers of unknown of primary: A meta-analysis. Eur. J. Cancer 2020, 127, 118–122.
- Ding, Y.; Jiang, J.; Xu, J.; Chen, Y.; Zheng, Y.; Jiang, W.; Mao, C.; Jiang, H.; Bao, X.; Shen, Y.; et al. Site-specific therapy in cancers of unknown primary site: A systematic review and meta-analysis. ESMO Open 2022, 7, 100407.
- Naing, A.; Meric-Bernstam, F.; Stephen, B.; Karp, D.D.; Hajjar, J.; Rodon Ahnert, J.; Piha-Paul, S.A.; Colen, R.R.; Jimenez, C.; Raghav, K.P.; et al. Phase 2 study of pembrolizumab in patients with advanced rare cancers. J. Immunother. Cancer 2020, 8, e000347.
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Comprehensive analysis of cancers of unknown primary for the biomarkers of response to immune checkpoint blockade therapy. Eur. J. Cancer 2018, 94, 179–186.
- Rassy, E.; Boussios, S.; Pavlidis, N. Genomic correlates of response and resistance to immune checkpoint inhibitors in carcinomas of unknown primary. Eur. J. Clin. Investig. 2021, 51, e13583.