Ovarian cancer (OC) is the most lethal gynaecological malignancy. The search for a widely affordable and accessible screening strategy to reduce mortality from OC is still ongoing. This coupled with the late-stage presentation and poor prognosis harbours significant health-economic implications. OC is also the most heritable of all cancers, with an estimated 25% of cases having a hereditary predisposition. Advancements in technology have detected multiple mutations, with the majority affecting the
BRCA1
and/or
BRCA2
genes. Women with
BRCA
mutations are at a significantly increased lifetime risk of developing OC, often presenting with a high-grade serous pathology, which is associated with higher mortality due to its aggressive characteristic.
1. Introduction
Globally, ovarian cancer (OC) is the eighth most common cancer among women and the eighteenth most common cancer overall
[1]. With around 7500 new cases every year and 5% of cancer-related deaths, OC is the sixth most common cancer as well as sixth most common cancer-related death among women in the United Kingdom (UK)
[2]. In the United States of America (USA), it is the fifth most common cancer-related death
[3]. OC has a lifetime risk of 1 in 78 and probability of mortality of 1 in 108
[2][3][2,3]. Overall, OC is the most lethal gynaecological malignancy and is recognised as the “silent killer” due to late-stage diagnosis caused by asymptomatic progression
[3].
Primary OC can be categorised into non-epithelial and epithelial, germ cell, and sex cord-stromal cancer, with epithelial being the most common. Non-epithelial accounts for approximately 10% of all OC and includes mainly germ cell tumours, sex cord-stromal tumours, and some extremely rare tumours
[4][5][4,5]. Germ cell tumours are extremely rare in menopausal women yet reported in the literature
[6]. Ovarian carcinosarcomas, accounting for only 1–4% of all OC, are composed of an epithelial as well as a sarcomatous component
[7]. Epithelial OC can be histologically categorised further into serous, clear cell, endometrioid, mucinous, or undifferentiated variety
[8]. Whilst the old line of thinking hypothesised that ovarian carcinogenesis arose from metaplasia of the ovarian surface epithelium into the various subtypes (serous, mucinous, clear cell, endometrioid, and transitional), a newer accepted theory by Kurman et al. provides a dualistic model
[9]. This classifies OC into Type I, which consists of clearly described precursor lesions, and Type II, where precursor lesions are not clearly described, wherein cancer may arise de novo from the tubal/ovarian epithelium. Type I, consisting of low-grade serous, mucinous, endometrioid, clear cell, and transitional cell carcinomas, is typically more indolent and presents at an earlier stage. Type II, consisting of high-grade serous carcinomas, undifferentiated carcinomas, and carcinosarcomas, behaves in a more aggressive manner, is genetically unstable, and typically presents at a later stage. Type II tumours have a high frequency of
TP53 mutations, whereas type I have specific mutations targeting cell signalling pathways, which in turn unsettle
BRCA expression, namely
KRAS,
BRAF,
ERBB2,
CTNNB1,
PTEN,
PIK3CA,
ARID1A, and
PPP2R1A [9][10][9,10]. Serous papillary peritoneal cancer shares common molecular, histological, and clinical features with epithelial OC, mainly high-grade serous, which made it reasonable to manage the two entities similarly
[11]. The recommended treatment of OC is radical surgery, followed by adjuvant chemotherapy. The therapeutic strategy of gestational OC depends on histology, stage, and gestational weeks
[12]. As the proteome closely mirrors the dynamic state of cells, tissues. and organisms, proteomics has great potential to deliver clinically relevant biomarkers for OC diagnosis and treatment
[13].
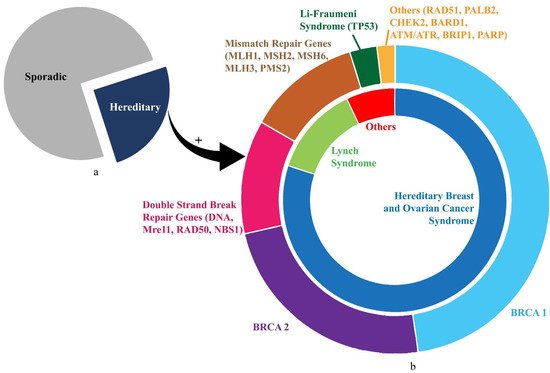
OC aetiology can be categorised into sporadic and hereditary. Approximately 23% of OC has a hereditary element, with the majority of those caused by defects within the BRCA DNA repair genes
[14]. It is estimated that the lifetime risk of OC is 40–50% amongst
BRCA1 mutation carriers and 20–30% in
BRCA2 mutation carriers, which is significantly higher than the general population
[15]. Several other genetic traits have been identified as risk factors for developing OC
[14][16][14,16]. These genetic factors offer an opportunity for primary and secondary prevention strategies to reduce the risk of these high-risk individuals developing OC. Platinum compounds and poly (ADP-ribose) polymerase (PARP) inhibitors are currently the two main classes of drugs active against cancer cells, harbouring DNA damage response and repair gene alterations
[17]. Genomic alterations in the DNA damage repair pathway are emerging as novel targets for treatment across different cancer types, especially OC, breast, and prostate cancer
[18][19][20][18,19,20].
Given the lack of evidence to support screening in high-risk individuals, a preventative approach, usually in the form of surgery or chemoprevention, is the first line of management in these women
[21][22][23][24][25][26][27][28][29][21,22,23,24,25,26,27,28,29]. Growing evidence suggests prevention strategies are both clinically and economically superior
[30][31][30,31]. However, whilst most evidence applies to sporadic OC, individuals at risk of hereditary ovarian cancers (HOC) may benefit even more from targeted interventions, which may be even more cost-effective in this high-risk population
[32][33][32,33].
2. Hereditary Ovarian Cancer (HOC)
HOC constitutes almost a quarter of all epithelial OC cases
[34]. Family history is the strongest risk. First- and second-degree relatives with OC carry a 3.6- and 2.9-fold lifetime risk of developing OC, respectively
[35].
HOC is also comprised of hereditary cancer syndromes (HCS), with mutations inherited in an autosomal dominant (AD) fashion leading to multiple primaries presenting at a young age. The two principal syndromes accounting for at least 20% of all epithelial OC are hereditary breast and ovarian cancer syndrome (HBOC) and Lynch syndrome (LS), also known as hereditary non-polyposis colorectal cancer syndrome (HNPCC)
[36][37][36,37].
HBOC constitutes approximately 80% of HOC and 15% of epithelial OC cases
[36]. Within HBOC, 65–85% of cases primarily stem from genomic mutations in
BRCA1 and
BRCA2 tumour-suppressor genes
[38]. These encode proteins for homologous recombination (HR) to repair DNA double-strand breaks (DSB) for maintaining genomic stability. The prevalence in the UK for
BRCA1 is thought to be 0.07–0.09% and for
BRCA2 0.14–0.22%
[39]. The lifetime risk for development and average age of onset of OC from
BRCA1 and
BRCA2 is 40% and 20% and 49 to 53 years and 55 to 58 years, respectively
[36][40][36,40]. More than 15% of HBOC cases arise due to mutations concerning other predisposition genes including
BARD1,
BRIP1,
CHEK2,
MRE11A,
MSH6,
NBN,
PALB2,
RAD50,
RAD51C, or
TP53 (
Figure 1).
BARD1,
BRIP1,
PALB2,
RAD50, RAD51C,
NBN, and
MRE11A gene mutations have been implicated in OC as part of the BRCA2/Fanconi anaemia signalling pathway in the event of nil
BRCA1 and/or
BRCA2 mutations. They constitute a significant portion of the DNA DSB repair machinery as well as next-generation sequencing (NGS)-based multigene panels
[41]. The
BARD1 gene showed one novel and three previously known genomic alterations in Ratajska et al.’s cohort of 255 unselected OC cases. These were almost nil in their control group, thus highlighting their pathogenic potential
[42]. The
BRIP1 mutation increases OC risk by 8-fold and decreases lifespan by almost four years
[43]. The
PALB2 gene mutation is prevalent in up to 4% of BRCA-negative HBOC cases. Yang et al. found a significant association between
PALB2 pathogenic variants and OC (i.e., a relative risk of almost three)
[44]. The
RAD51C gene is prevalent in up to 2.9% of HBOC families negative for
BRCA1 and/or
BRCA2 mutations. Meindl et al. found six pathogenic variants in
RAD51C among 1100 German families, yielding a relative risk of six for developing OC
[45].
RAD50 and
MRE11A are constituents of the MRE11 complex. Heikkinen et al. identified germline mutations in
RAD50 and
MRE11A among 151 HBOC families
[46]. Meanwhile, Ramus et al. revealed that the prevalence of germline mutations of the
NBN gene was very low (0.2%), hence not contributing significantly to OC risk
[47]. In epithelial OC, DNA mismatch repair (MMR) deficiency is the second most common cause of HOC—only behind HR deficiency—accounting for 10–15% of HOC
[48]. Furthermore, it has been reported that high mRNA levels of
MSH6,
MLH1, and
PMS2 were associated with a prolonged overall survival in OC. That supports the potential positive prognostic value of MMR genes in OC patients treated with platinum-based chemotherapy.
Figure 1. The illustration shows distribution of gene mutations in ovarian cancer. (a) Proportion of sporadic (75%) and hereditary (25%) ovarian cancer cases. (b) Details of HOC. The inner circle shows the proportions contributed by HBOC and Lynch Syndrome. The outer circle demonstrates the divisions shared by prominent genetic mutations (BRCA1, BRCA2, TP53) and mutation groups (mismatch repair genes, double-strand-break repair genes) corresponding approximately to the syndromic association in the inner circle.
LS is the second commonest cause of HOC responsible for approximately 15% cases and 4% of epithelial OC cases
[38]. It primarily involves colorectal cancer along with an increased frequency of extracolonic tumours, including endometrial, ovarian, urogenital, brain, renal, gastric, and biliary. The lifetime risk of developing OC with LS is approximately 8 to 12%, and the mean age of presentation is about 43 years. Mutations in MMR genes have been implicated in LS, namely
MSH2 (38% cases),
MLH1 (32%),
PMS2 (15%), and
MSH6 (15%)
[49]. Grindedal et al. noted a 30-year OC survival of 71.5% in 144 MMR mutation carriers with OC, seemingly better than
BRCA mutation survival
[50].
Other rarer syndromes concerning HOC include Li–Fraumeni syndrome, Cowden syndrome, Peutz–Jeghers syndrome, diffuse gastric cancer syndrome, and neurofibromatosis type 1 syndrome. These arise primarily due to mutations in the
TP53,
PTEN,
STK11,
CDH1, and
NF1 genes, respectively
[51].
Figure 1 illustrates the inherited gene mutations in HOC.