Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Cláudio Maia.
Prostate cancer (PCa) is the second most diagnosed cancer and the fourth leading cause of cancer-related death in men in the Western world. Delineation of pathogenetic pathways and key driver molecular alterations involved in PCa development has provided a roadmap for the evaluation of biomarkers in predicting disease outcome and to identify potential therapeutic targets. Chemotherapeutic agents introduced from the 1990s include the taxanes (paclitaxel, docetaxel, and cabazitaxel), which are the anticancer drugs used most frequently for PCa treatment.
- PCa
- taxane-based drugs
- transmembrane proteins
- combination therapy
1. Onset and Development of PCa
The human prostate gland is the major accessory gland of the male reproductive system, located frontal to the rectum and immediately below the urinary bladder, surrounding the prostatic urethra and the ejaculatory ducts [11,12][1][2]. Normal prostate tissue consists of prostatic ducts lined with epithelial cells surrounded by fibromuscular stroma [13,14][3][4]. Homeostasis of normal prostate tissue is maintained by the crosstalk between epithelial cells and the surrounding stromal components [15,16][5][6]. The glandular prostatic epithelium is a well-organized tissue composed of acini and ducts consisting of three types of cells: luminal, basal, and neuroendrocrine cells (Figure 1). Luminal cells are columnar epithelial cells specialized in the production of prostatic secretions, including prostate-specific antigen (PSA), and are responsible for the main prostate function [17][7]. Basal cells adhere to the basement membrane and have the ability to produce several components essential to the maintenance of cell growth [18,19][8][9]. Neuroendocrine cells comprise less than 1% of the prostatic epithelium and express chromogranin A, synaptophysin, enolase 2, and CD56, which promote the growth of the prostate [20][10]. Interactions between the epithelium and basement membrane are fundamental to maintain epithelial cell polarity involving apical and basal surfaces, which represent the well-differentiated cell state [13][3]. The non-epithelial tissue of the prostate, referred to as the stroma, is composed essentially by fibroblasts, smooth muscle cells, and extracellular matrix (ECM) proteins (Figure 1) [15][5]. The ECM forms a dynamic and structured mixture of collagens, proteoglycans, thrombospondin, and hyaluronic acid, which responds to tissue injuries and allows its regeneration [16][6].

Figure 1. Schematic representation of the proposed model of the cellular events associated with the development and progression of PCa. The prostate epithelium is composed of the luminal cells responsible for the production of prostatic secretions and the basal cells that are on the base of the epithelium in contact with the basement membrane. Located among the epithelial cells also exist neuroendocrine cells that are involved in the regulation of secretory activity and prostate cell growth. Prostate epithelial cells maintain contact with the stroma, including smooth muscle cells, fibroblast cells, and components of the extracellular matrix (ECM). Damage in the prostate normal epithelium induces the development of pre-neoplastic lesions called prostatic intraepithelial neoplasia (PIN). This stage progresses to localized prostate adenocarcinoma where the basal cell layer is lost, which then becomes invasive adenocarcinoma when the basement membrane is degraded, and neoplastic cells can invade the lymphatic system and other organs including the liver, lungs, and bones.
Considering the onset of PCa, there is good agreement that this cancer develops from prostate epithelial cells [14][4]. However, conflicting evidence exists regarding if the oncogenic transformation in PCa arises from basal [19,21][9][11] or luminal epithelial cells [22,23][12][13]. In addition, it also has been hypothesized that PCa arising from luminal cells is more aggressive than that arising from basal cells [21][11]. The prostatic epithelium can be damaged and drive the carcinogenesis of the prostate due to several factors, including inflammation, infections, genetic/epigenetic changes, persistent activation by androgens, exposure to carcinogens, and/or genetic factors [14,24][4][14]. The first identifiable histologic alteration in prostate malignant transformation is so-called prostatic intraepithelial neoplasia (PIN) (Figure 1) [25][15].
Prostatic adenocarcinoma mostly arises in the peripheral zone of the prostate and initially is represented as a small foci of intraductal dysplasia, that with time differentiates and progresses into an invasive adenocarcinoma (Figure 1) [30][16]. The tumor foci lead to a disruption of prostate tissue and a decrease in glandular activity and prostatic fluid production [31][17]. Histologically, PCa is characterized by the destruction of the basal cell layer, derangement of the basement membrane, decreased epithelial cell polarity, and lack of connection of the glandular acini formed by the prostate epithelial cells [32][18]. As the tumor progresses, neoplastic cells increase the production of proteolytic enzymes, which cause degradation of the basement membrane, allowing the spread to adjacent tissues and the development of a metastatic disease [33][19]; firstly, to the lymph nodes and then to distant organs, including the bones, liver, and lungs, with bone as the most common site of metastasis [34][20].
Androgens play a central role in the control of the normal prostate, as well as PCa cell growth and proliferation [14][4]. Androgens are the primary regulators of the proliferation/apoptosis ratio, stimulating proliferation and inhibiting apoptosis of prostate cells, and, thus, inducing the development of PCa [14,36][4][21]. The major circulating androgen, testosterone, can be converted into DHT by the activity of 5α-reductase enzyme. Both testosterone and DHT exert their actions through binding to the AR. PCa growth and disease progression is initially dependent on AR activation. The main mechanism of action leads to the nuclear translocation of the ligand–receptor complex and subsequent binding to the androgen response elements (AREs), which initiates the transcription of genes that regulate cellular differentiation, proliferation, and apoptosis (Figure 2) [27,36,37][21][22][23].
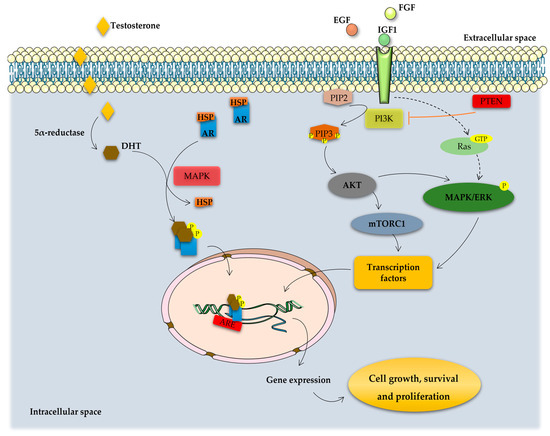
Figure 2. Overview of the molecular pathways associated with the development of CRPC. In the cytoplasm, activity of AR is regulated by ligand binding and heat-shock proteins (HSP). Testosterone is transported into the cytoplasm of androgen-receptive cells and is converted to 5α-dihydrotestosterone (DHT) by the enzyme 5α-reductase. DHT binding leads to dissociation of AR from HSP and its phosphorylation by the mitogen-activated protein kinase (MAPK), which is followed by receptor dimerization and translocation into the nucleus, where it binds to the androgen response elements (AREs) in the DNA, activating the transcription of genes essential for cell growth, survival, and proliferation. On the other hand, PCa cell fate is controlled by receptor tyrosine kinases (RTKs) activated by several growth factors, such as insulin-like growth factor (IGF1), fibroblast growth factor (FGF), and epidermal growth factor (EGF). RTK activation leads to the stimulation of phosphatidylinositol 3-kinase (PI3K) that phosphorylates phosphatidylinositol 4,5-bisphosphonate (PIP2) into phosphatidylinositol 3–5-triphosphate (PIP3). This process is inhibited by the tumor suppressor phosphatase and tensin homolog (PTEN). PIP3 activates, which subsequently removes the inhibition on the mTOR/Raptor complex (also known as mTORC1), thus leading to mTORC1 activation. mTORC1 is pivotal in the translation of proteins for protein synthesis and activation of transcription factors that translocate to the nucleus, inducing the expression of pro-proliferation and anti-apoptotic genes. Other intracellular pathways also converging on the mTORC1 complex are constituted by the Ras-dependent pathway. Activated Ras (a small GTPase) phosphorylates and activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascade, regulating the activity of several transcription factors that are important for the cell cycle and proliferation. The activation of these signaling pathways inhibits apoptosis and induces the proliferation, invasion, and migration of PCa cells, also being implicated in tumor metastization.
In primary PCa, the action of AR retains the same role as in a normal prostate; for example, synthesis of PSA and modulating lipid metabolism [22][12]. However, it also triggers other events that promote epithelial cell growth, such as the induction of the type II transmembrane serine protease (TMPRSS2):ETS fusion [26,38][24][25]. The TMPRSS2 is an androgen-regulated gene overexpressed in PCa, which encodes a protein belonging to the serine protease family that functions in prostate carcinogenesis and relies on gene fusion with ETS transcription factors, such as the ETS-related gene (ERG) and ETV1. The TMPRSS2:ETS fusion is considered the most common chromosomal rearrangement in PCa and drives the overexpression of ETS oncogenes, previously identified as the most expressed proto-oncogenes present on malignant epithelial prostate cells [38,39,40][25][26][27].
Patients that acquire resistance to the use of androgen-deprivation therapy (ADT) inevitably develop CRPC, a more lethal form of PCa. The role of AR in PCa progression and development of CRPC has been attributed to several factors, such as AR gene amplification, activating mutations, and aberrant expression of co-activators [37,43,44][23][28][29]. These alterations lead to an increased AR expression, activation of AR by non-androgenic ligands, broadened ligand specificity and sensitivity, and increased AR transactivation, which ultimately contribute to tumor cell growth in a low androgen-environment [36,44,45][21][29][30]. AR mutations in primary PCa are rare, but these mutations are prevalent in about 50% of CRPC [46,47][31][32]. These mutations lead to alterations that improve the functional activity of the receptor, such as increased AR sensitivity to low levels of ligand, non-androgen ligand binding, ligand-independent activation, and AR-independent pathways [41,46,47][31][32][33].
Various growth factors, cytokines, kinases, and other proteins have been shown to interact with and activate AR in a ligand-independent manner, including insulin-like growth factor (IGF1), fibroblast growth factor (FGF), and epidermal growth factor (EGF) [51,52][34][35]. These growth factors activate tyrosine receptor kinases, which results in the activation of phosphatidylinositol 3-kinase (PI3K) and subsequently the PI3K/AKT pathway (Figure 2) [53][36].
2. Current Use of Chemotherapy in PCa
Treatment approaches for PCa differ depending on the stage of the disease. Several types of therapeutic options are available, such as surgery, cryosurgery, radiation therapy, hormone therapy, chemotherapy, vaccine treatment, immunotherapy, and bone-directed treatment [56][37]. Active surveillance is the recommended treatment option for low-risk PCa, monitoring its progression while not undergoing definitive therapy [57][38]. Therapeutic approaches based on surgery often are used in combination with therapeutic approaches based on drugs, namely hormone therapy and chemotherapy. As the non-neoplastic prostate cells, PCa cells need androgens to grow and survive, making ADT an effective first-line therapy. This therapy can involve two approaches: surgical castration (i.e., orchiectomy) or, more commonly, chemical castration with drugs targeting AR signaling regulated by the hypothalamic pituitary gonadal axis (e.g., GnRH agonists, AR antagonists, and CYP17A1 inhibitors). This castration reduces tissue androgen levels and also reduces the expression of several androgen-regulated genes [34][20].
As the disease progresses to the CRPC stage, treatment involves the use of chemotherapeutic drugs (Figure 3). Mitoxantrone was the first cytotoxic chemotherapy approved by the FDA for metastatic PCa [60][39]. Next, other therapeutic agents for the treatment of CRPC were included, such as the chemotherapeutic taxanes paclitaxel and docetaxel. After the discovery of the mechanism of action of paclitaxel, which involves tubulin binding and enhanced microtubule polymerization resulting in mitotic arrest [61][40], other taxanes were explored, and their synthetic and semisynthetic analogues with the best properties and improved water solubility were produced [62][41]. The most successful semisynthetic analogue of paclitaxel is docetaxel, which is a taxane derivative that induces microtubule stabilization, arresting cells in the G2/M phase of the cell cycle, and it induces bcl-2 phosphorylation, promoting a cascade of events that leads to apoptotic cell death (Figure 4) [63][42].
Figure 3. Chemical structures of chemotherapy drugs, namely paclitaxel, docetaxel, and cabazitaxel (image extracted by PubChem on 13 July 2023).
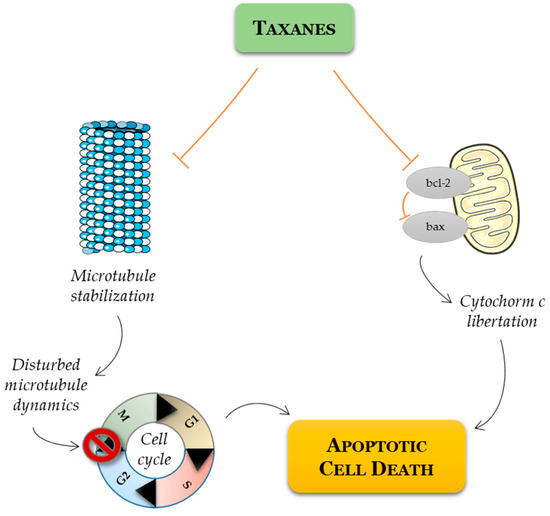
Figure 4. Schematic representation of mode of action of taxanes on cancer cell. Taxanes have been described as exerting their antitumor efficacy via distinct modes of action: mitotic and apoptotic action. Taxanes bind to microtubules and thereby prevent their disassembly, resulting in G2/M cell cycle arrest and apoptosis. Alternatively, taxanes may inhibit the expression of antiapoptotic Bcl-2, favoring apoptotic cell death through the relief of BAX-mediated cytochrome c release.
Multiple prospective randomized clinical trials have been designed to evaluate the efficacy and toxicity of therapies, and diverse combinations have been attempted [71,72,73][43][44][45]. The CHAARTED (Chemohormonal Therapy versus Androgen Ablation Randomized Trial for Extensive Disease in PCa) and STAMPEDE (Systemic Therapy in Advancing or Metastatic PCa: Evaluation of Drug Efficacy) trials showed a remarkable overall survival benefit when combining ADT with docetaxel, as well as increased time to progression to castration-resistant status [74,75][46][47]. In the FIRSTANA (Cabazitaxel Versus Docetaxel Both With Prednisone in Patients With Metastatic CRPC) trial, cabazitaxel showed no superiority versus docetaxel for overall survival of PCa patients as a first-line treatment [76][48]. Although docetaxel and cabazitaxel have similar efficacy, there are differences in their toxicity profiles. Low doses of cabazitaxel are associated with lesser overall toxicity than docetaxel [77][49].
It is evident that taxanes are constantly being upgraded both in terms of mechanistic and clinical aspects, and their success in the treatment of PCa (castration-sensitive and castration-resistant settings) lies in the continued development of rational combination therapy strategies with the explicit goal of improving overall survival [73][45].
43. Transmembrane Proteins as a Potential Therapeutic Target in Combination with Taxanes
A transmembrane protein is a type of protein located either in the lipid bilayer of the plasma membrane or in the membrane of organelles [81][50]. Different from monotopic proteins, transmembrane protein structure completely crosses the membrane [82][51]. Representing approximately 30% of the genome, transmembrane proteins are essential for many cellular processes [83][52]. These proteins are responsible for cell–cell and cell–environment communication, through signal transduction, the binding of receptors to hormones and neurotransmitters, and the transport of substances across the membrane [82,83][51][52]. There are two types of transmembrane proteins regarding their structure: either alpha-helical proteins or beta-barrel proteins.
4.1. MDR1
3.1. MDR1
The efflux pump Multidrug Resistance Protein 1 (MDR1), also called p-glycoprotein, is a protein composed of 12 transmembrane domains and a single monomer of 170 kDa [84][53]. This protein is part of the ATP-binding cassette (ABC) transporter family and is encoded by the p-glycoprotein (ABCB1) gene, located in the region 7q21 [84][53]. The overexpression of MDR1 has been shown to be partially responsible for drug resistance in PCa, due to higher drug efflux [85][54]. Regarding p-glycoprotein expression, Kawai et al. reported that both PCa and normal prostate epithelial cells are positive for the expression of the MDR1 gene [86][55]. Using monoclonal antibodies to detect the presence of p-glycoprotein, the same study confirmed that this protein is asymmetrically expressed in the inner and outer zones of nonmalignant prostate glands [86][55]. To investigate whether the presence of p-glycoprotein in blood exosomes could be a marker to diagnose docetaxel resistance in PCa, Kato et al. tested the susceptibility to docetaxel and cabazitaxel drugs in parental and docetaxel-resistant PC3 cell lines considering p-glycoprotein expression [87][56]. It was demonstrated that docetaxel-sensitive PC3 cells showed little or no expression of this protein, while docetaxel-resistant PC3 cells showed high expression of p-glycoprotein [87][56]. The knockdown of the ABCB1 gene was also performed in docetaxel-resistant PC3 cells. The results indicated an improvement in docetaxel sensitivity when compared with the negative control. These findings confirm the relationship between p-glycoprotein expression and docetaxel resistance [87][56].4.2. MRP4
3.2. MRP4
Similarly to the MDR1 protein, the MRP4 protein, also known as multidrug resistance protein 4, is part of the ABC transporter family [90][57]. This transmembrane protein is present in almost all tissues in the body, such as the brain, kidney, liver, erythrocytes, platelets, adrenal gland, and pancreas [91][58]. MRP4 is responsible for the transportation of prostaglandins E1 and E2 (PGE1 and PGE2), as well as cAMP and cGMP [92][59]. The MRP4 protein was reported as being highly overexpressed in docetaxel-resistant C4-2B cells, while no expression of MRP4 was detected in docetaxel-sensitive C4-2B cells [93][60]. To assess if the overexpression of MRP4 leads to docetaxel resistance, a combined treatment of MRP4 knockdown plus docetaxel exposure was applied to the docetaxel-resistant C4-2B cell line. The results showed diminished cell viability, indicating a re-sensitization to docetaxel treatment [93][60].4.3. CD44
3.3. CD44
CD44 is a non-kinase cell surface transmembrane glycoprotein [95][61]. This important hyaluronate receptor is overexpressed in cancer stem cells and is involved in cellular adhesion and communication, lymphopoiesis, myelopoiesis, and angiogenesis [95][61]. In regard to cancer, CD44 is implicated in metastasis, cellular growth, proliferation, migration, and invasion [95][61]. There are several isoforms for the CD44 protein and some of them have been associated with PCa, namely the CD44s, CD44v6, and CD44v7-10 isoforms [95][61]. Furthermore, CD44 is also overexpressed in this type of cancer and is associated with aggressive biological behavior and a poor prognosis [95][61]. CD44 expression is upregulated by transforming growth factor-beta 1 (TGF-β1) in PCa cells [95][61]. CD44 is expressed in PC3 cells and it was demonstrated that this receptor regulates glucose metabolism, intracellular reactive oxygen species (ROS), and cell proliferation in these cells; however, CD44 is not expressed in LNCaP cells [90][57]. Collected data also point to the regulation of proliferation, invasion, and migration via PDK1 and PFKFB4, which are enzymes that regulate glucose metabolism and are modulated by CD44 [96][62]. To investigate whether the presence of p-glycoprotein in blood exosomes could be a marker to diagnose docetaxel resistance in PCa, Kato et al. tested the susceptibility to docetaxel and cabazitaxel drugs in parental and docetaxel-resistant PC3 cell lines considering p-glycoprotein expression [87][56]. It was demonstrated that docetaxel-sensitive PC3 cells showed little or no expression of this protein, while docetaxel-resistant PC3 cells showed high expression of p-glycoprotein [87][56]. The knockdown of the ABCB1 gene was also performed in docetaxel-resistant PC3 cells. The results indicated an improvement in docetaxel sensitivity when compared with the negative control. These findings confirm the relationship between p-glycoprotein expression and docetaxel resistance [87][56].4.2. MRP4
Similarly to the MDR1 protein, the MRP4 protein, also known as multidrug resistance protein 4, is part of the ABC transporter family [90]. This transmembrane protein is present in almost all tissues in the body, such as the brain, kidney, liver, erythrocytes, platelets, adrenal gland, and pancreas [91]. MRP4 is responsible for the transportation of prostaglandins E1 and E2 (PGE1 and PGE2), as well as cAMP and cGMP [92]. The MRP4 protein was reported as being highly overexpressed in docetaxel-resistant C4-2B cells, while no expression of MRP4 was detected in docetaxel-sensitive C4-2B cells [93]. To assess if the overexpression of MRP4 leads to docetaxel resistance, a combined treatment of MRP4 knockdown plus docetaxel exposure was applied to the docetaxel-resistant C4-2B cell line. The results showed diminished cell viability, indicating a re-sensitization to docetaxel treatment [93].4.3. CD44
3.4. CD133
CD44 is a non-kinase cell surface transmembrane glycoprotein [95]. This important hyaluronate receptor is overexpressed in cancer stem cells and is involved in cellular adhesion and communication, lymphopoiesis, myelopoiesis, and angiogenesis [95]. In regard pentaspan to cancer, CD44 is implicated in metastasis, cellular growth, proliferation, migration, and invasion [95]. Thansmere are several isoforms for the CD44 protein and some of them have been associated with PCa, namely the CD44s, CD44v6, and CD44v7-10 isoforms [95]. Furthermore, CD44 is bralso overexpressed in this type of cancer and is associated with aggressive biological behavior and a poor prognosis [95]. CD44 expression is upregulated be gly transforming growth factor-beta 1 (TGF-β1) in PCa cells [95]. CD44 is exopressed in PC3 cells and it was demonstrated that this receptor regulates glucose metabolism, intracellular reactive oxygen species (ROS), and cell proliferation in these cells; however, CD44 is not expressed in LNCaP cells [90]. Collected data also point to the regulation of proliferation, invasion, and tein promigration via PDK1 and PFKFB4, which are enzymes that regulate glucose metabolism and are modulated by CD44 [96].4.4. CD133
The pentaspan transmembraine glycoprotein prominin-1, also known as CD133, is a protein mostly found in the microvilli of different epithelial cells but is also expressed in numerous types of cancer, such as breast, ovarian, and PCa and other non-epithelial cell types [98,99][63][64]. CD133 is frequently used as a biomarker for the detection of cancer stem cells [99][64]. The molecular function of this glycoprotein has not been yet fully clarified, but there is strong evidence pointing towards a role in membrane organization, due to its preferred location on the microvilli, and a role in spermatozoa biogenesis and photoreceptor disc formation [98][63]. Regarding the photoreceptor disc formation, it is known that a mutation on the CD133 gene is the cause of a type of macular degeneration called Stargardt disease [98][63]. CD133 is also important in angiogenesis through the regulation of the expression of vascular endothelial growth factor (VEGF) [98][63].4.5. SLCO1B3
3.5. SLCO1B3
Belonging to the Solute Carriers superfamily, SLCO1B3, also called organic anion-transporting polypeptide (OATP) [102][65], is a sodium-independent transporter of both endogenous substrates, such as bilirubin, bile salts, steroid conjugates, bromosulfophthalein (BSP), and Taurocholate (TCA) [102[65][66],103], and exogenous substrates, such as antihistamines, blood-glucose-lowering drugs, statins, heart medications, and docetaxel and paclitaxel [102,104][65][67]. Konig et al. confirmed that, under normal conditions, SLCO1B3 is exclusively expressed in hepatocytes, with its subcellular location on the basolateral plasma membrane of those cells [105][68]. Additionally, a preferred lobular zonation was also observed, where the hepatocytes near the central vein showed a higher expression of this protein when compared to other locations within the liver [105][68].4.6. EGFR
3.6. EGFR
The transmembrane glycoproteins epidermal growth factor receptor (EGFR), together with HER-2/neu (erbB-2), HER-3 (erbB-3), and HER-4 (erbB-4), belong to the HER (erbB) family of membrane receptors (Figure 5) [111][69]. All these receptors are expressed in both normal and malignant cells, playing important roles in cell proliferation and differentiation [112][70]. All four family members have a very similar structure, consisting of three regions: the first is an extracellular ligand-binding region, which, in the case of EGFR, is the binding region for the epidermal growth factor (EGF), transforming growth factor-a (TGF-a), amphiregulin (AR), Heparin-binding EGF-like growth factor (HB-EGF), and betacellulin (BTC) [111,112][69][70]. HER2 dimerizes with EGFR [113][71] and has no exclusive natural ligand [111][69]. The second region, a transmembrane domain, consists of a single hydrophobic anchor sequence that crosses the cell membrane only once [112][70]. Lastly, the third region acts as a binding site for intracellular substrates, and therefore activates signaling pathways [112][70]. The intracellular domain has tyrosine kinase activity [111][69].4.7. STEAP1
3.7. STEAP1
STEAP1, together with STEAP2-4, is part of the six-transmembrane epithelial antigen of prostate (STEAP) family of proteins [119][72]. The STEAP1 protein is overexpressed in several human cancers, including prostate, bladder, colon ovary, breast, and cervical cancer [120][73]. Although its function remains unclear, some studies have pointed out that STEAP1 is involved in metal reductase activity, and also in the transport of ions such as Na+, Ca2+, and K+ [121][74]. STEAP1 is highly expressed in LNCaP cells and also at significant levels in the C4-2B cell line [122][75]. Regarding the effect of STEAP1 knockdown in LNCaP cells, reduced cell viability was observed in comparison to the control group [123][76]. This result was supported by the cell proliferation index, showing a 0.3-fold decrease in LNCaP cells knocked down for STEAP1. In addition to its effect on the inhibition of cell proliferation, the STEAP1 knockdown increased the number of apoptotic cells [123][76].4.8. CCL2/CCR2
3.8. CCL2/CCR2
A member of the CC beta chemokine family, monocyte chemoattractant protein 1 (MCP-1 or CCL2) is a monomeric polypeptide [124,125][77][78]. CCL2 is an agonist for its main receptor, the transmembrane protein CCR2, and also an agonist for the CCR4 and CCR5 receptors [125][78]. Moreover, other chemokines act as agonists on the CCR2 receptor, such as CCL7 and CCL8; therefore, there is an overlap of ligands and receptors [125][78]. It is found in many cell types, such as endothelium, epithelium, and bone marrow; CCL2’s main function is recruiting immune cells, such as monocytes, T lymphocytes, and natural killer (NK) cells [124,125][77][78]. It has been demonstrated that in the tumor–bone microenvironment of metastasis collected from patients diagnosed with PCa, several cytokines were upregulated, namely CCL2, which was expressed four times more on the tumor than on the normal tissue adjacent to the tumor [126][79]. Furthermore, a correlation between CCL2 serum levels and PCa progression can be found, indicating that elevated CCL2 serum levels are associated with bone metastasis [127][80]. The genetic variation found in CCL2 also supports its role in cancer progression and development; three SNPs of the CCL2 gene are linked to higher Gleason scores [128][81]. Regarding in vitro culture of PCa cells, it is known that several cell lines expressed different levels of the CCR2 receptor, but PC3 and VcaP cell lines showed the highest levels of expression [126][79]. CCR2 expression has also been correlated to the Gleason score and pathological stage [124,127][77][80]. There is evidence pointing towards CCL2 as having an important role in cell migration. Using PC3 cells, the treatment with human recombinant CCL2 (hrCCL2) showed that cells present higher migration compared to control, and that the effect is dose-dependent [126][79]. In the same way, the presence of either an anti-CCR5 neutralizing antibody or anti–human CCL2 and anti–mouse CCL2/JE neutralizing antibodies led to a decrease in cell migration [126][79]. Furthermore, in PC3 cells, Akt phosphorylation is stimulated in a dose-dependent manner by CCL2 [126][79]. To investigate the results of CCL2 inhibition in PCa, several assays were performed using the DU145 cell line [129][82]. Three experimental groups were delineated: a chemosensitive cell line (DU145), a paclitaxel-resistant cell line (DU145- TxR), and a paclitaxel- and cabazitaxel-resistant cell line (DU145- TxR/CxR). Using cDNA microarray analysis data, it was confirmed that CCL2 gene expression was 70-fold higher in DU145-TxR cells when compared to DU145 cells, and 43-fold higher in DU145-TxR/CxR cells [129][82]. The level of CCL2 in the cell medium was also measured, indicating a higher level in the DU145- TxR/CxR cell culture and the lowest in the DU145 cell culture [129][82]. The exposure to cabazitaxel was not able to alter the CCR2 receptor expression in any cell line [129][82].4.9. VEGFR
3.9. VEGFR
The Vascular Endothelial Growth Factor (VEGF) family of ligands and its receptors (VEGFR) are responsible for the development of both blood and lymphatic vascular networks [134][83]. The receptors are VEGFR1, VEGFR2, and VEGFR3. All receptors are comprised of an extracellular domain containing seven immunoglobulin homology domain repeats, a transmembrane domain, and a tyrosine kinase domain [134][83]. Lu et al. engineered a novel anti-VEGFR2 fully human Ab and applied it to PCa [137][84]. Using a PC3 xenograft PCa model, researchers assessed the therapeutic potential of this Ab in comparison to ramucirumab and docetaxel [137][84]. Mice were divided into the following treatment options: ramucirumab, anti- VEGFR2, docetaxel, anti-VEGFR2 plus docetaxel, or ramucirumab plus docetaxel. Already approved by the FDA for gastrointestinal stromal tumors, Sunitinib is an oral multi-tyrosine kinase inhibitor, which includes an effect against VEGFR2 [135][85]. Studies on PCa cell lines demonstrated that Sunitinib had an antitumor effect that was dose- and time-dependent [135][85]. A phase I/II clinical trial was also performed using Sunitinib in a combined treatment with docetaxel and prednisone [138][86]. Cediranib (AZD-2171) is an inhibitor of all three VEGFRs, and its effect is being studied in several types of cancer, including PCa [140][87]. In the PC3 and DU145 cell lines, Cediranib as a sole treatment decreased cell survival, induced apoptosis, and cell motility [140][87]. Cediranib has already reached clinical studies. In a phase I study, AZD2171 was administrated to 19 patients with CRPC [143][88]. Unfortunately, none of them achieved a ≥50% decline in PSA levels [143][88]. A phase II trial also included CRPC patients, who had already been exposed to docetaxel, and their disease continued to progress [138][86].54. Conclusions
Taxane-based chemotherapeutic drugs are currently the main approach when it comes to PCa treatment. Even though this type of therapy has good results in improving patient survival, the development of resistance to chemotherapeutic drugs remains a great obstacle.References
- Lee, C.H.; Akin-Olugbade, O.; Kirschenbaum, A. Overview of prostate anatomy, histology, and pathology. Endocrinol. Metab. Clin. N. Am. 2011, 40, 565–575.
- Ittmann, M. Anatomy and Histology of the Human and Murine Prostate. Cold Spring Harb. Perspect. Med. 2018, 8, a030346.
- Leong, K.G.; Wang, B.-E.; Johnson, L.; Gao, W.-Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808.
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta 2016, 1863 Pt A, 1238–1260.
- Corn, P.G. The tumor microenvironment in prostate cancer: Elucidating molecular pathways for therapy development. Cancer Manag. Res. 2012, 4, 183–193.
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510.
- Yadav, N.; Heemers, H.V. Androgen action in the prostate gland. Minerva Urol. Nefrol. 2012, 64, 35–49.
- Long, R.M.; Morrissey, C.; Fitzpatrick, J.M.; Watson, R.W. Prostate epithelial cell differentiation and its relevance to the under-standing of prostate cancer therapies. Clin. Sci. 2005, 108, 1–11.
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615.
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285.
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nature 2013, 15, 274–283.
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal Cells Are Favored as the Cell of Origin for Prostate Cancer. Cell Rep. 2014, 8, 1339–1346.
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500.
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000.
- Ayala, A.G.; Ro, J.Y. Prostatic intraepithelial neoplasia: Recent advances. Arch. Pathol. Lab. Med. 2007, 131, 1257–1266.
- Schiebler, M.L.; E Tomaszewski, J.; Bezzi, M.; Pollack, H.M.; Kressel, H.Y.; Cohen, E.K.; Altman, H.G.; Gefter, W.B.; Wein, A.J.; Axel, L. Prostatic carcinoma and benign prostatic hyperplasia: Correlation of high-resolution MR and histopathologic findings. Radiology 1989, 172, 131–137.
- Schiebler, M.L.; Schnall, M.D.; Pollack, H.M.; Lenkinski, R.E.; E Tomaszewski, J.; Wein, A.J.; Whittington, R.; Rauschning, W.; Kressel, H.Y. Current role of MR imaging in the staging of adenocarcinoma of the prostate. Radiology 1993, 189, 339–352.
- Ulmert, D.; O’Brien, M.F.; Bjartell, A.S.; Lilja, H. Prostate kallikrein markers in diagnosis, risk stratification and prognosis. Nat. Rev. Urol. 2009, 6, 384–391.
- Duffy, M.J. The role of proteolytic enzymes in cancer invasion and metastasis. Clin. Exp. Metastasis 1992, 10, 145–155.
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140.
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238.
- Murray, T.B.J. The Pathogenesis of Prostate Cancer. In Prostate Cancer; Exon Publications: Brisbane, Australia, 2021; pp. 29–42.
- Culig, Z.; Klocker, H.; Bartsch, G.; Hobisch, A. Androgen receptors in prostate cancer. Endocr. Relat. Cancer 2002, 9, 155–170.
- Joshua, A.; Evans, A.; Van der Kwast, T.; Zielenska, M.; Meeker, A.; Chinnaiyan, A.; Squire, J. Prostatic preneoplasia and beyond. Biochim. Biophys. Acta 2008, 1785, 156–181.
- Knuuttila, M.; Mehmood, A.; Mäki-Jouppila, J.; Ryberg, H.; Taimen, P.; Knaapila, J.; Ettala, O.; Boström, P.J.; Ohlsson, C.; Venäläinen, M.S.; et al. Intratumoral androgen levels are linked to TMPRSS2-ERG fusion in prostate cancer. Endocr.-Relat. Cancer 2018, 25, 807–819.
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 409–436.
- Wei, T.; Lu, J.; Ma, T.; Huang, H.; Kocher, J.-P.; Wang, L. Re-Evaluate Fusion Genes in Prostate Cancer. Cancer Inform. 2021, 20, 11769351211027592.
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23.
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54.
- Devlin, H.-L.; Mudryj, M. Progression of prostate cancer: Multiple pathways to androgen independence. Cancer Lett. 2009, 274, 177–186.
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894.
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926.
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15.
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478.
- Zhu, M.-L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr.-Relat. Cancer 2008, 15, 841–849.
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507.
- Schatten, H. Brief Overview of Prostate Cancer Statistics, Grading, Diagnosis and Treatment Strategies. Adv. Exp. Med. Biol. 2018, 1095, 1–14.
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA—J. Am. Med. Assoc. 2017, 317, 2532–2542.
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C.M.; et al. Chemotherapy with mitoxantrone plus prednisone or pred-nisone alone for symptomatic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996, 14, 1756–1764.
- Long, H.J. Paclitaxel (Taxol): A Novel Anticancer Chemotherapeutic Drug. Mayo Clin. Proc. 1994, 69, 341–345.
- Kubník, J.; Pavlíčková, V.; Ruml, T.; Rimpelová, S. Current Perspectives on Taxanes: Focus on Their Bioactivity, Delivery and Combination Therapy. Plants 2021, 10, 569.
- Pienta, K.J. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin. Oncol. 2001, 28 (Suppl. 15), 3–7.
- Van Soest, R.J.; De Wit, R. Irrefutable evidence for the use of docetaxel in newly diagnosed metastatic prostate cancer: Results from the STAMPEDE and CHAARTED trials. BMC Med. 2015, 13, 304.
- Damodaran, S.; Lang, J.M.; Jarrard, D.F. Targeting Metastatic Hormone Sensitive Prostate Cancer: Chemohormonal Therapy and New Combinatorial Approaches. J. Urol. 2019, 201, 876–885.
- Huebner, N.A.; Shariat, S.F.; Resch, I.; Gust, K.; Kramer, G. The role of taxane-based chemotherapy in the treatment of prostate cancer. Curr. Opin. Urol. 2020, 30, 527–533.
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177.
- Clarke, N.W.; Ali, A.; Ingleby, F.C.; Hoyle, A.; Amos, C.L.; Attard, G.; Brawley, C.D.; Calvert, J.; Chowdhury, S.; Cook, A.; et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: Long-term survival results from the STAMPEDE trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1992–2003.
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial—FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197.
- Eisenberger, M.; Hardy-Bessard, A.-C.; Kim, C.S.; Géczi, L.; Ford, D.; Mourey, L.; Carles, J.; Parente, P.; Font, A.; Kacso, G.; et al. Phase III Study Comparing a Reduced Dose of Cabazitaxel (20 mg/m2) and the Currently Approved Dose (25 mg/m2) in Postdocetaxel Patients With Metastatic Castration-Resistant Prostate Cancer—PROSELICA. J. Clin. Oncol. 2017, 35, 3198–3206.
- Schmit, K.; Michiels, C. TMEM Proteins in Cancer: A Review. Front. Pharmacol. 2018, 9, 1345.
- Ryu, H.; Fuwad, A.; Yoon, S.; Jang, H.; Lee, J.C.; Kim, S.M.; Jeon, T.-J. Biomimetic Membranes with Transmembrane Proteins: State-of-the-Art in Transmembrane Protein Applications. Int. J. Mol. Sci. 2019, 20, 1437.
- Marx, S.; Dal Maso, T.; Chen, J.W.; Bury, M.; Wouters, J.; Michiels, C.; Le Calvé, B. Transmembrane (TMEM) protein family members: Poorly charac-terized even if essential for the metastatic process. Semin. Cancer Biol. 2020, 60, 96–106.
- Bossennec, M.; Di Roio, A.; Caux, C.; Ménétrier-Caux, C. MDR1 in immunity: Friend or foe? Oncoimmunology 2018, 7, e1499388.
- Ganju, A.; Yallapu, M.M.; Khan, S.; Behrman, S.W.; Chauhan, S.C.; Jaggi, M. Nanoways to overcome docetaxel resistance in prostate cancer. Drug Resist. Updat. 2014, 17, 13–23.
- Kawai, K.; Sakurai, M.; Sakai, T.; Misaki, M.; Kusano, I.; Shiraishi, T.; Yatani, R. Demonstration of MDR1 P-glycoprotein isoform expression in benign and malignant human prostate cells by isoform-specific monoclonal antibodies. Cancer Lett. 2000, 150, 147–153.
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum exosomal P-glycoprotein is a potential marker to di-agnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol. Oncol. 2015, 33, e15–e385.
- Hardy, D.; Bill, R.M.; Jawhari, A.; Rothnie, A.J. Functional Expression of Multidrug Resistance Protein 4 MRP4/ABCC4. SLAS Discov. 2019, 24, 1000–1008.
- Ravna, A.W.; Sager, G. Molecular model of the outward facing state of the human multidrug resistance protein 4 (MRP4/ABCC4). Bioorg. Med. Chem. Lett. 2008, 18, 3481–3483.
- Sauna, Z.E.; Nandigama, K.; Ambudkar, S.V. Multidrug resistance protein 4 (ABCC4)-mediated ATP hydrolysis: Effect of transport substrates and characterization of the post-hydrolysis transition state. J. Biol. Chem. 2004, 279, 48855–48864.
- Li, Y.-F.; Ji, H.-H.; Zhang, Z.-L.; Zhang, T.-T.; Gan, W.; Zhang, S.-F. Targeting MRP4 expression by anti-androgen treatment reverses MRP4-mediated docetaxel resistance in castration-resistant prostate cancer. Oncol. Lett. 2017, 14, 1748–1756.
- Mesrati, M.H.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11, 1850.
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151.
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269.
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18.
- Sun, R.; Ying, Y.; Tang, Z.; Liu, T.; Shi, F.; Li, H.; Guo, T.; Huang, S.; Lai, R. The Emerging Role of the SLCO1B3 Protein in Cancer Resistance. Protein Pept. Lett. 2020, 27, 17–29.
- Liu, S.; Peng, T.; Wang, Z.; Li, Y.; Zhang, H.; Gui, C. Effect of rare coding variants of charged amino acid residues on the function of human organic anion transporting polypeptide 1B3 (SLCO1B3). Biochem. Biophys. Res. Commun. 2021, 557, 1–7.
- Pressler, H.; Sissung, T.M.; Venzon, D.; Price, D.K.; Figg, W.D. Expression of OATP Family Members in Hormone-Related Cancers: Potential Markers of Progression. PLoS ONE 2011, 6, e20372.
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and Genomic Organization of a New Hepatocellular Organic Anion Transporting Polypeptide. J. Biol. Chem. 2000, 275, 23161–23168.
- Bellezza, I.; Bracarda, S.; Caserta, C.; Minelli, A. Targeting of EGFR tyrosine kinase by ZD1839 (“Iressa”) in androgen-responsive prostate cancer in vitro. Mol. Genet. Metab. 2006, 88, 114–122.
- Rude Voldborg, B.; Damstrup, L.; Spang-Thomsen, M.; Skovgaard Poulsen, H. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206.
- Jathal, M.K.; Steele, T.M.; Siddiqui, S.; Mooso, B.A.; D’abronzo, L.S.; Drake, C.M.; Whang, Y.E.; Ghosh, P.M. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. Br. J. Cancer 2019, 121, 237–248.
- Xu, M.; Evans, L.; Bizzaro, C.L.; Quaglia, F.; Verrillo, C.E.; Li, L.; Stieglmaier, J.; Schiewer, M.J.; Languino, L.R.; Kelly, W.K. STEAP1–4 (Six-Transmembrane Epithelial Antigen of the Prostate 1–4) and Their Clinical Implications for Prostate Cancer. Cancers 2022, 14, 4034.
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587.
- Zhao, C.; Xiong, K.; Ji, Z.; Liu, F.; Li, X. The Prognostic Value and Immunological Role of STEAP1 in Pan-Cancer: A Result of Da-ta-Based Analysis. Oxid. Med. Cell Longev. 2022, 2022, 8297011.
- Rocha, S.M.; Nascimento, D.; Coelho, R.S.; Cardoso, A.M.; Passarinha, L.A.; Socorro, S.; Maia, C.J. STEAP1 Knockdown Decreases the Sensitivity of Prostate Cancer Cells to Paclitaxel, Docetaxel and Cabazitaxel. Int. J. Mol. Sci. 2023, 24, 6643.
- Gomes, I.M.; Rocha, S.M.; Gaspar, C.; Alvelos, M.I.; Santos, C.R.; Socorro, S.; Maia, C.J. Knockdown of STEAP1 inhibits cell growth and induces apoptosis in LNCaP prostate cancer cells counteracting the effect of androgens. Med. Oncol. 2018, 35, 40.
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing an-tibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424.
- Kadomoto, S.; Izumi, K.; Mizokami, A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8530.
- Loberg, R.D.; Day, L.L.; Harwood, J.; Ying, C.; John, L.N.S.; Giles, R.; Neeley, C.K.; Pienta, K.J. CCL2 is a Potent Regulator of Prostate Cancer Cell Migration and Proliferation. Neoplasia 2006, 8, 578–586.
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of the MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 2009, 26, 161–169.
- Sun, T.; Mary, L.G.-S.; Oh, W.K.; Freedman, M.L.; Pomerantz, M.; Pienta, K.J.; Kantoff, P.W. Inherited Variants in the Chemokine CCL2 Gene and Prostate Cancer Aggressiveness in a Caucasian Cohort. Clin. Cancer Res. 2015, 17, 1546–1552.
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019, 110, 279–288.
- Secker, G.A.; Harvey, N.L. Regulation of VEGFR Signalling in Lymphatic Vascular Development and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 7760.
- Lu, R.; Chiu, C.; Liu, I.; Chang, Y.; Liu, Y.; Wu, H. Novel human Ab against vascular endothelial growth factor receptor 2 shows therapeutic potential for leukemia and prostate cancer. Cancer Sci. 2019, 110, 3773–3787.
- Park, H.S.; Hong, S.K.; Oh, M.M.; Yoon, C.Y.; Jeong, S.J.; Byun, S.S.; Cheon, J.; Lee, S.E.; Moon, D.G. Synergistic antitumor effect of NVP-BEZ235 and sunitinib on docetaxel-resistant human castration-resistant prostate cancer cells. Anticancer. Res. 2014, 34, 3457–3468.
- Adesunloye, B.A.; Karzai, F.H.; Dahut, W.L. Angiogenesis Inhibitors in the Treatment of Prostate Cancer. Chem. Immunol. Allergy 2014, 99, 197–215.
- Momeny, M.; Sankanian, G.; Hamzehlou, S.; Yousefi, H.; Esmaeili, F.; Alishahi, Z.; Karimi, B.; Zandi, Z.; Shamsaiegahkani, S.; Sabourinejad, Z.; et al. Cediranib, an inhibitor of vascular endothelial growth factor receptor kinases, inhibits proliferation and invasion of prostate adenocarcinoma cells. Eur. J. Pharmacol. 2020, 882, 173298.
- Ryan, C.J.; Stadler, W.M.; Roth, B.; Hutcheon, D.; Conry, S.; Puchalski, T.; Morris, C.; Small, E.J. Phase I dose escalation and pharmacokinetic study of AZD2171, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinase, in patients with hormone refractory prostate cancer (HRPC). Investig. New Drugs 2007, 25, 445–451.
More