Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shane Whelan and Version 2 by Sirius Huang.
Urinary tract infections (UTIs) are among the most common bacterial infections, especially among women and older adults, leading to a significant global healthcare cost burden. Uropathogenic Escherichia coli (UPEC) are the most common cause and accounts for the majority of community-acquired UTIs. Infection by UPEC can cause discomfort, polyuria, and fever. More serious clinical consequences can result in urosepsis, kidney damage, and death. UPEC is a highly adaptive pathogen which presents significant treatment challenges rooted in a complex interplay of molecular factors that allow UPEC to evade host defences, persist within the urinary tract, and resist antibiotic therapy.
- uropathogenic E. coli
- biofilm formation
- antibiotic resistance
1. Introduction
Urinary tract infections are one of the most common bacterial infections, with an estimated 400 million cases and 230,000 deaths worldwide in 2019 [1]. Some 50% of women will experience a UTI at least once in their lifetime, with recurrence being common [2]. Furthermore, UTIs are one of the most common infections reported in older adults, being second only to respiratory infections in hospitalised patients over 65 [3]. The economic burden of UTIs is high, representing 6% of medical visits in the United States and costing approximately USD 1.6 billion annually [4]. A European study which sought to determine the cost associated with complicated urinary tract infection in eight countries with a high prevalence of multidrug-resistant Gram-negative bacteria found that the average cost of treatment was EUR 5700 per case [5]. An Irish study from 2014 estimated the primary care cost of UTI in Ireland to be EUR 19.2 million annually [6]. UTI infections fall into distinct clinical categories as defined by the European Association of Urology (EAU), which in turn guides treatment [7]. Uncomplicated UTIs are described as infections involving non-pregnant, pre-menopausal women with no known relevant urological abnormalities or comorbidities, while complicated UTIs are those affecting men, pregnant women, patients with anatomical or functional abnormalities, renal disease, indwelling urinary catheters and those with certain immunosuppressive co-morbidities [7]. A recurrent UTI (R-UTI) is defined as two or more uncomplicated or complicated UTI infections in six months, or three infections within one year that are new infections with separate organisms [8]. Certain conditions or abnormalities may predispose a person to recurrent infection. This can occur due to relapse and infection by the same persistent organism following unsuccessful treatment, but in the majority of cases it results from infection with a separate organism, post the resolution of symptoms [9].
The main cause of UTIs is infection by uropathogenic Escherichia coli (UPEC), responsible for 75% of uncomplicated UTIs and 65% of complicated UTIs [10]. UPEC form part of a group of pathogenic E. coli known as extraintestinal pathogenic E. coli (ExPEC), a collection of four pathotypes that are each classified by their respective site of isolation, and have adopted virulence mechanisms that allow them to proliferate and cause disease in host sites outside of the intestinal tract [11][12][11,12]. ExPEC are separable from diarrheagenic E. coli (DEC), a group of six E. coli pathotypes which cause intestinal infections, and each possess a different combination of virulence characteristics [13]. Infection begins when bacteria that reside in the gut enter the urethra and colonise epithelial cells and eventually migrate upwards to colonise the bladder. The initiation of the host’s innate immune response to infection represents a complex cascade of events. Central to this response are pattern recognition receptors (PRRs). Notably among this family are Toll-like receptors (TLRs), which recognise pathogen-associated molecular patterns (PAMPs) [14]. TLRs are expressed across a number of cell types, including immune cells such as NK cells, macrophages, and leukocytes, as well as non-immune cells such as bladder, ureter, and kidney urothelial cells [15][16][15,16]. The 10 TLRs (TRL1-10) which are found in humans are distinguishable by the ligands they recognise and the resulting signalling pathways they activate [17]. Critical to the host’s immune response during invasion by UPEC is TLR4, the sensing receptor for Gram-negative lipopolysaccharide (LPS) [18]. Upon detection of LPS, TLR-4 activates the intracellular signalling pathway NF-κB which regulates the expression of many proinflammatory cytokines and chemokines as well as leukocyte attracting chemoattractants [19][20][19,20]. Further inflammation is stimulated by TLR-5, which responds to the presence of bacteria flagellin [21]. While the locally attracted phagocytes are effective at clearing extracellular pathogens, TLR-4 additionally mediates the expulsion of intracellular pathogens through cAMP-dependent exocytosis from bladder epithelial cells [22].
2. Virulence Factors
The ability of UPEC to colonise the bladder and resist antibiotic therapy is attributed to several virulence factors, many of which are also related to biofilm formation [23][24][25][26,58,59]. These include adhesion factors such as Type 1 fimbriae, P fimbriae, curli fibres, S fimbriae, F1C fimbriae, Dr fimbriae, afimbrial adhesins, and PapC [26][27][60,61]. These adhesion factors enable UPEC to adhere to and invade host cells and tissues, evade the host immune response, and colonise the urinary tract [28][29][62,63]. In addition, the extracellular matrix of UPEC biofilms is composed mainly of amyloid curli fibres, which are encoded by the curli operon [30][64]. Curli fibres play a crucial role in UPEC biofilm formation and the extracellular matrix they compose provides protection from environmental stresses such as antibiotics, host defences, and nutrient deprivation [31][65]. Toxins such as α-hemolysin, cytotoxic necrotizing factor, and serine protease autotransporter can cause tissue damage, proteolytically cleave immune cell complexes, and trigger morphological changes in host cells which facilitate bacterial persistence [32][33][34][34,35,36].
Moreover, the ability of UPEC to acquire iron is highly dependent on siderophore-iron transporter proteins [35][33]. These proteins enable UPEC to scavenge iron from the host and other sources, which is essential for bacterial growth and survival in the environment of the urinary tract where iron levels are infinitesimal [35][33]. A representative set of the virulence factors that can be expressed by UPEC are shown in Figure 1.
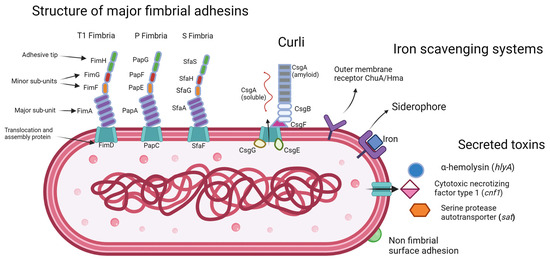
Figure 1.
E. coli
with a representative set of virulence factors of the UPEC pathotype.
Virulence factors such as adhesions and siderophores can coalesce into pathogenicity islands, be mobilised in transposons, and be transferred on plasmids [36][66]. This transfer of virulence factors enables UPEC to adapt and evolve rapidly in response to environmental pressures and facilitates the spread of antibiotic resistance genes among interspecies bacterial populations.
2.1. Adhesions
2.1.1. Type 1 Fimbriae
Fimbria adhesions are extracellular protein structures that allow UPEC to attach to bladder cells and resist expulsion from the urinary system through the flow of urine. The best defined and most common fimbria adhesions are type 1fimbria (T1F). Genomic analysis of UPEC populations have reported prevalence of T1F between 86–100% [26][37][38][60,67,68]. Apart from initial attachment, T1F have been shown to mediate the invasion of bladder and uroepithelial cells, forming IBCs which can form persistent reservoirs of infection, are more resistant to antibiotics, and are speculated to contribute to recurrent infections [39][40][69,70].
T1F are encoded by the fim operon found in the majority of UPEC, but importantly, is not always expressed [41][71]. This operon contains nine genes, fimA to fimI with fimA expressing the main structural element, and fimC and fimD playing roles in the transport and assembly of the fimbriae [42][72]. fimH is responsible for facilitating binding of the fimbriae to host receptors, and has specificity for D-mannose-containing structures on host cells, often uroplakins [43][23]. The D-mannose-specific structures required for attachment are more numerous in the lower urinary tract than in the kidneys, so it was long believed that T1F do not play a role in pyelonephritis; however, more recent data suggest that T1F are still expressed in kidney infections, though they play the role of facilitating inter-bacterial adhesion and biofilm formation, rather than attachment and host cell invasion [44][73].
Expression of T1F is controlled by a reversible process called phase variation; a process by which the expression of bacterial surface factors, primarily adhesions, can be switched on or off [45][74]. Phase variation, or antigenic variation, was first described as it related to T1F and can be considered a method of host immune evasion, allowing bacteria to lower its expression of antigenic surface adhesions when switched off as well as to prevent coexpression and interference between surface components [46][47][75,76]. Here, the process is controlled by an invertible DNA element containing the promoter sequence for fimA. This sequence, which contains two inverted repeats flanking an intervening DNA segment, lines up with fimA to allow site specific recombination mediated by a recombinase enzyme and promotes transcription [48][77]. Expression through phase variation is thought to be regulated through a complex network of systems which responds to stress, and are likely to become activated during infection because of the hostile environment of the host urinary tract exerting pressure on invading UPEC [49][78]. The gene regulation which occurs during this response is regulated by a system of small-nucleotide alarmones, pGpp, ppGpp, and pppGpp (guanosine tetra- and pentaphosphates) and is produced by the enzymes RelA and SpoT [50][51][79,80]. Higher intrinsic levels of (p)ppGpp in E. coli result in high alarmone concentration, and this system activates the regulator FimB that controls the phase variation of type 1 fimbriae in E. coli [52][81].
2.1.2. P Fimbriae
Like T1F, P fimbriae are extracellular protein appendages which play a role in the adherence of UPEC to uroepithelial cells [53][25]. They differ from type 1 fimbriae in that their adherence is mannose resistant, binding to globoseries glycosphingolipids as opposed to the mannose dependent adherence of type-1 fimbriae to mono-mannose structures [54][24]. They are also more associated with upper urinary tract infections and pyelonephritis (P fimbriae for pyelonephritis fimbriae) and are considered a particularly important virulence factor in UPEC, as they mediate attachment to epithelial cells in the kidneys and renal tubules [44][55][73,82]. P fimbriae are encoded chromosomally by the pap operon. The main fimbriae structure is encoded by papA, papEF encode adapter subunits and papG encodes the terminal adhesive protein [53][25]. Because of this, the papG gene is usually used as a molecular marker to determine the prevalence of P fimbriae among UPEC. Studies have reported the prevalence of the pap operon to be 14–28% in UPEC causing cystitis, 5–7% in commensal E. coli and 71–91% of UPEC isolated from patients with pyelonephritis [55][56][82,83]. Importantly, papG has four molecular variants, with distinct binding specificities. It follows therefore, that some of the molecular variants of papG create more virulent strains of UPEC for certain hosts, and several studies have sought to characterise the differences between these variants and the diseases they are associated with. For example, PapGII has been encountered more than other alleles in isolates from women with pyelonephritis, while PapGIII is found more commonly in UPEC which is causative of cystitis [57][58][84,85]. PapGI and PapGIV are encountered rarely and are less understood than the other two molecular confirmations. UPEC which are positive for the papGII gene has also been shown to be positive for additional virulence factors, which may contribute to its association with more severe, upper UTI [57][84].
Regulation of the pap operon by phase variation involves the formation of differential methylation patterns resulting from the competition between DNA adenine methylase (Dam) and leucine-responsive regulatory protein (Lrp) for binding to two sets of overlapping binding sites within the pap regulatory region [59][86]. The regulatory region is composed of two genes, the first is papI which activates the on-phase by interacting with Lrp-pap DNA complexes when a specific GATC sequences is methylated by Dam [60][87]. The second is papB which both activates papI transcription but also represses transcription when protein concentrations are high [61][88].
2.1.3. S Fimbriae
S fimbriae are mannose-resistant adhesions which are encoded by the sfa operon. This operon consists of nine genes. The SfaA protein forms the dominant sub-unit, while SfaG, SfaH, and SfaS form minor sub-units, and SfaF forms the outer membrane translocation assembly protein [62][63][89,90]. Regulatory proteins SfaB and SfaC control expression, and like other fimbriae are manged by phase variation, that in this case, is influenced by temperature, osmolarity, glucose concentration, and other environmental factors [64][91]. S fimbriae has specificity for alpha-sialyl-2,3-alpha-galactose residues present on the glycoproteins of urothelial tissues in the bladder and kidneys [62][65][89,92]. Apart from urinary tract infections, S fimbriae-positive E. coli have been known to cause meningitis in new-borns, likely due to their binding potential for sialo-glycoproteins which are also found on brain micro vascular endothelial cells [66][93]. Studies which used molecular analysis to determine the prevalence of the sfa operon in E. coli have reported figures between 42–87% [26][37][38][67][60,67,68,94]. The sfa operon has been found more commonly in strains which form strong biofilms in vitro, as determined by tissue culture plate methods [67][68][94,95].
2.1.4. The Afa/Dr Family of Fimbrial Surface Adhesins
The Afa/Dr family of adhesions are particularly associated with UPEC that cause cystitis in children, as well as pyelonephritis and recurrent urinary tract infections in young and pregnant women [69][70][71][96,97,98]. It is also regularly found in the genome of ST131, a particularly virulent UPEC clone [72][99]. Dr fimbria (diffuse adherence fimbria) are part of the Afa/Dr family of fimbrial surface adhesions and are separable from other adhesions by the wide range of binding specificity they possess. They can bind, firstly to the Dr blood group antigen on decay-accelerating factor (DAF), which is a glycoprotein present on the surface of uroepithelial cells [73][100]. Secondly, Dr fimbria can recognise carcinoembryonic antigen (CEA)-related cell adhesion molecules (CEACAMs) CEA, CEACAM1, CEACAM3, and CEACAM6 as binding receptors [74][101]. Interactions between Dr fimbria and these receptors have been shown to mediate internalisation and invasion of UPEC into the host epithelial cells [74][75][101,102].
The genes encoding afa/dr adhesions are composed of a primary transcriptional unit containing afaA which is a regulatory gene, afaB which encodes a chaperone protein, afaC which encodes an usher protein, afaE which encodes a major adhesion subunit, and a tip capping subunit protein encoded by afaD. This gene cluster is accompanied by a separate minor transcriptional gene; afaF [69][96]. Phase mediated expression of the afa/dr family adhesions is, like P fimbriae, mediated by differential methylation patterns caused by Dam and Lrp interactions in the afa/dr promoter region [76][103]. Activation, however, is carried out by integration host factor (IHF) and repression is mediated by H-NS, a histone-like nucleoid structuring protein [72][99]. Previously it has been shown that insertion sequences (IS) elements when inserted into the regulatory region of the Afa/dr operon cause increased expression in a ST131 UPEC clone [72][99].
2.1.5. Non-Fimbrial Adhesins
Repeat-in-toxins (RTX) are a family of proteins usually transported to the cell surface through the type I secretory system [77][104]. They include toxins such as cytolysins and hemolysin, but also non-fimbrial adhesions [78][105]. Type one secretion A (tosA) is one such non fimbrial adhesion, encoded by the tosRCBDAEF operon [79][106]. Expressed on the cell surface, this adhesion can bind to human kidney and epithelial cells in the upper urinary tract [79][106]. The operon is regulated by TosR, with assistance from the global regulatory proteins H-NS and Lrp and its expression involves cross talk with the regulatory systems of other fimbrial adhesions, including Pap and Foc, allowing for cell surface organisation of both fimbrial and non-fimbrial adhesions [80][107]. Over expression of TosA has been shown to increase biofilm formation in vitro, in both LB broth and in human urine [80][107].
2.2. Curli
Curli is a functional amyloid protein that is an essential component of the biofilm matrix in many bacterial species, including E. coli [81][108]. Curli fibres are formed by the self-assembly of the major curli subunit protein, CsgA, which polymerises into fibrils on the bacterial cell surface upon contact with the minor subunit protein CsgB [82][109]. Curli production is regulated by the transcription factor CsgD, which activates the expression of the curli biosynthesis genes, which include csgA, csgB, and csgG, as well as genes involved in nucleotide biosynthesis and other metabolic pathways [31][65]. CsgE, CsgF, and CsgG proteins comprise the outer membrane curli secretion system, with CsgG forming a pore like outer membrane structure and CsgE and CsgF functioning as chaperones for secreted curli fibers [83][110].
The curli biosynthesis pathway in E. coli is particularly sensitive to changes in nucleotide biosynthesis, as disruption of the de novo purine biosynthetic pathway leads to a reduction in the intracellular level of cyclic-di-GMP, a secondary messenger molecule that is required for the activation of the csgD gene [84][111]. Cyclic-di-GMP is synthesised by diguanylate cyclases and degraded by phosphodiesterases, and its intracellular level is tightly regulated by a network of enzymes and signalling pathways [85][112]. The disruption of purine biosynthesis leads to a decrease in the intracellular level of cyclic-di-GMP, which in turn leads to a reduction in curli production and biofilm formation [30][64]. A study which created transposon mutants found that a strain with an insertion in the purL gene, resulted in a knockout variant which had reduced curli production and diminished biofilm formation [86][113]. The purL gene codes for formyl-glycinamide ribonucleotide amido-transferase (FGAR-AT), which is an enzyme that catalyses the fourth step of the purine biosynthetic pathway [87][114].
The involvement of the CsgD regulator in extracellular matrix production induced by extracellular nitrate suggests that the regulation of biofilm formation is also linked to the availability of nutrients in the bacterial environment. This was demonstrated by the creation of mutant strains lacking the membrane bound nitrate reductases NarGHJI and NarZYWV and produced increased biofilm as a result [88][115].
2.3. Biofilm Formation
Biofilm formation is a complex process that involves various genes and regulatory mechanisms. The fimbrial genes fimB, fimE, and the curli assembly component csgE are crucial for biofilm formation at all stages. However, during the initial 12 h of formation, genes related to gene transcription and motility play a more significant role along with these adhesion genes [89][116]. Several studies have investigated the relationship between phylogenetic groups and biofilm formation. Phylogroups B2 and D are most frequently associated with stronger biofilm formation or exhibit multidrug resistance and carry a high percentage of fimbrial, iron uptake, and toxin genes [90][91][117,118]. Moreover, one study reported no correlation between phylogenetic classification and biofilm formation, but a correlation was observed between antibiotic resistance and strong biofilm formation irrespective of phylogroup [52][81]. More generally, the biofilm formation of UPEC is variable between clonally distinct strains, and can include both weak and strong formers [92][119].
A study which characterised multidrug-resistant UPEC by phylogenic group found that adhesion genes were more commonly found in group B2 and C, and these strains had a higher resistance to fluoroquinolones, trimethoprim-sulfamethoxazole, and amoxicillin-clavulanate [93][30]. Among adhesin genes, the prevalence of fimH was the highest (91.8%), followed by pap (79.3%), sfa (12.0%), and afa (7.7%). Additionally, biofilm formation was significantly correlated with the pap adhesion gene [93][30].A study from Iran reported statistically higher levels of papEF in strong biofilm formers, and the adhesion genes fimH, focG, sfaS, set-1, and cvaC were more common in weaker biofilm formers [94][31]. Researchers have previously shown that the biofilm forming tendencies of UPEC isolates are variable and not apparent from colony morphology, and that sub-inhibitory concentrations of ciprofloxacin and trimethoprim can, in some cases induce UPEC isolates to form stronger biofilms in vitro, even when resistant to antibiotics [92][95][119,120].
2.4. Toxin Production and Cytotoxic Effects
2.4.1. Hemolysin
α-hemolysin (HlyA) is pore forming toxin and virulence factor produced by many strains of UPEC. The prevalence of hemolysin production by UPEC is between 21–47% of isolates, as determined by multiple studies [96][97][98][99][121,122,123,124]. The operon which encodes α-hemolysin consists of four genes, hlyCABD [100][101][125,126]. HlyA can trigger apoptosis in host cells by modulating cell death pathways and interfering with cell process regulators. For example it has been shown that protein kinase B (PBK) which inhibits apoptosis and stimulates immune responses in host cells, is itself inhibited by HlyA in human bladder epithelial cells [102][127]. The immune response is further disrupted because HlyA inhibits the metabolic enzyme ACLY, which results in low acetyl-CoA levels. This modulates histone acetylation and prevents the adequate promotion of proinflammatory cytokine and chemokine genes [103][128]. During infection, HlyA stimulates the upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) which causes macrophage accumulation and kidney damage during episodes of acute pyelonephritis [32][34]. UPEC isolates that were found to be stronger biofilm formers in vitro have been shown to have higher hemolysin activity [97][122].
2.4.2. Cytotoxic Necrotizing Factor Type 1 (CNF1)
Many strains of UPEC produce cytotoxic necrotizing factor type 1 (CNF1), an endotoxin that modulates the activity of host cells by interacting with the Rho family of GTPases and restricting their activation [104][129]. These GTPases act as molecular switches that control many cellular processes, such as the formation of actin structures, cell motility, cell cycle progression, and phagocytosis [105][106][107][130,131,132]. A range of effects on human and animal cell lines have been documented after exposure to CNF toxins, including elongation and multinucleation, which in vivo will lead to host cell damage and bacterial persistence [33][35]. There are four known variants of CNF: CNF1, CNF2, CNF3, and CNFY. They each work by deamidating Rho Q63, and the resulting inhibition of GTPase leads to a reorganisation of actin stress fibres, modification of the host cytoskeleton, and cell damage [108][109][133,134]. CNF2 differs from the chromosomally mediated CNF1 and CNF3 in that it is plasmid encoded [110][135].
CNF1 is encoded by the gene cnf1, which has been found in a pathogenicity island that also contains genes encoding P-fimbriae and α-hemolysin [111][136]. CNF1 internalises into host cells via receptor-mediated endocytosis and contains two distinct binding regions, one located on the N terminus composed of amino acids 135 to 164, and one on the C terminus composed of amino acids 683 to 730, the host receptor being laminin receptor precursor protein (LRP), which is expressed on the surface of eukaryotic cells [112][113][137,138].
Epidemiological data suggest that CNF1 is much more common in UPEC as compared to commensal E. coli isolates, and in mouse models it has been shown that CNF1 positive strains more extensively colonise the bladder, are recovered from the urine in greater numbers, and were better able to resist killing by human neutrophils than CNF1 negative strains [114][115][139,140]. CNF1 is also associated with E. coli strains that are causative of neonatal meningitis, as well as diarrhoea in children [116][117][141,142]. Regarding meningitis, it is thought that E. coli producing CNF1 can more effectively cross the blood–brain barrier and infect the brain, and has been shown to increase disease severity in a mouse model of meningitis [118][119][143,144].
2.4.3. Serine Protease Autotransporter
SPATE (serine protease autotransporters of Enterobacteriaceae) are a subgroup of secreted proteins called autotransporters that contribute to the survivability and virulence of UPEC [120][145]. Among this family, the secreted autotransporter toxin encoded by the sat gene has particular importance to UPEC virulence due to its cytotoxic activity on urinary tract epithelial cells [121][146]. First discovered in E. coli CFT073, a strain isolated from a patient with acute pyelonephritis, it shows in vitro cytopathic activity in kidney and bladder cell lines and is found significantly more frequently in E. coli associated with pyelonephritis than in faecal E. coli strains [121][146]. When investigating the kidney damage caused by sat in a mouse model of UTI, Guyer et al. (2002) found that sat induces vacuolation in glomeruli and proximal tubules, a histological change that has been seen in other toxins such as VacA toxin of Helicobacter pylori [122][123][147,148]. Sat has also been shown to exhibit immunomodulatory effects, presenting proteolytic effects against complement proteins that would otherwise destroy invading bacteria once they enter the bloodstream following endothelial infection [34][36].
Table 1 shows virulence factors related to fimbrial and non-fimbrial adhesions with their corresponding genes and host cell receptors, as well as toxins produced by UPEC with associated genes and the corresponding effects on host cells.
Table 1.
Adhesions and toxins in UPEC with associated genes and host interactions.
| Adhesions | Genes | Host Receptor |
|---|---|---|
| Type 1 Fimbriae | fimABCDEFGHI [42][72] | D-mannose-containing receptors, uroplakins [43][23] |
| P Fimbriae | papABCDEFG [57][84] | mannose-resistant binding to globoseries glycosphingolipids [54][24] |
| Afa/Dr | afaABCDEF [69][96] | decay-accelerating factor, carcinoembryonic antigens [73][74][100,101] |
| S Fimbriae | sfaABCDEFGHS [124][149] | Sialo-glycoproteins [62][65][89,92] |
| Type one secretion A | tosRCBDAEF [79][106] | human kidney and epithelial cells [79][106] |
| Toxins | Genes | Effect in host |
| α-hemolysin | hlyCABD [100][101][125,126] | PBK and ACLY inhibition, GM-CSF upregulation [32][102][103][34,127,128] |
| Cytotoxic necrotizing factor | cnf1 [111][136] | Inhibits Rho family of GTPases [104][129] |
| Serine protease autotransporter | sat [121][146] | Induces vacuolation in glomeruli and proximal tubules, degrades complement proteins [34][122][36,147] |
2.5. Iron Acquisition Systems
Metal ions are essential nutrients required by UPEC for many crucial biological processes, including the formation of enzymes involved in cellular metabolism [125][150]. These are metalloenzymes, which require metals including iron, nickel, zinc and copper as co-factors, with iron being the most commonly required [126][151]. As iron is a key factor in the proliferation of UPEC within the urinary tract, and the retention of iron to deprive invading pathogens of the nutrient composes part of the defensive strategy on the part of the host, acquisition of iron is a key point of competition between host and pathogen [127][128][129][152,153,154]. UPEC have evolved several iron acquisition systems in response to this competition, the first being iron-chelating molecules called siderophores, of which there are four that UPEC can synthesis and secrete, those being enterobactin, salmochelin, yersiniabactin, and aerobactin [130][155]. Of these, enterobactin is the most conserved among clinical E. coli isolates, with the others appearing in various combinations [130][131][155,156]. These variations in genetic carriage of siderophore systems appear to be an important part of the UPEC immune evasion strategy, as host mechanisms can prevent iron acquisition activity of specific siderophores, for example, the innate immune protein lipocalin-2 (Lcn2) prevents enterobactin activity by binding ferric and aferric enterobactin [132][157]. Enterobactin is encoded by the operon entABCDEF; salmochelin is encoded by the genes iroB and iroE; yersinianactin is encoded by ybtSETU, irp1, and irp2; and aerobactin is encoded by iucABCD. The presence of irp2 and iuc has previously been reported to be most prevalent in serogroups 025, 015 and 08 [133][52]. Rezatofighi et al. (2021) has reported that siderophores genes fyuA and iutA are found more frequently in group B2 than in other phylogroups [134][158] While iutD has been reported to be highly distributed into groups B2 and D among multidrug-resistant UPEC (MDR-UPEC) and extensively drug resistant UPEC (XDR-UPEC) [91][118]. In addition to siderophores, UPEC also utilise host haemoglobin to acquire iron using the outer membrane receptors ChuA and Hma [135][136][159,160]. Strains lacking either of these receptors has been shown to be ineffective colonisers of the kidneys in a mouse model of UTI, indicating their importance for successful colonisation of the upper urinary tract [136][160]. Even among asymptomatic bacteriuria strains, which lack many of the common virulence factors expressed by other UPEC, they will express iron acquisition systems, showing their necessity in urinary tract colonisation [137][161].
Fimbrial adhesions, curli, and non fimbrial surface adhesions are expressed on the cell surface to aid in colonisation and persistence through UPEC and host cell interactions, facilitating intra-cellular invasion as well as cell aggregation and biofilm formation. The expression of these surface proteins is regulated by phase variation. Outer membrane receptors and siderophores sequester iron which is essential for many cell processes and a scarce resource in the urinary tract. Secreted toxins cause tissue damage in an infected host and assist in immune evasion.