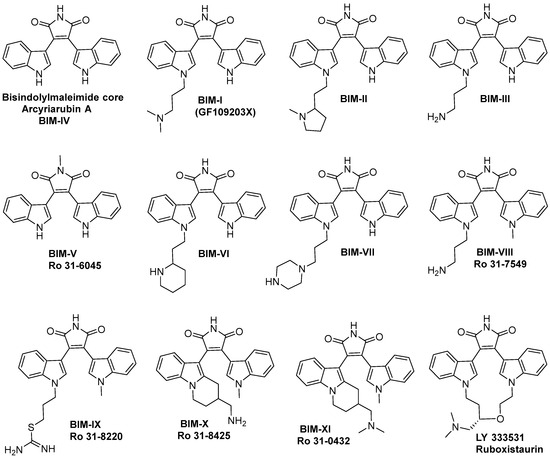
Bisindolylmaleimide (BIM)-type compounds arise from natural sources such as arcyriarubin and are biosynthetically related to indolocarbazoles. BIMs are commonly the immediate synthetic precursors of indolocarbazoles, lacking a central bond between the two aromatic units and making them more flexible and drug-like. Synthetic endeavours within this class of compounds are broad and have led to the development of both remarkably potent and selective protein kinase inhibitors. Clinical BIM examples include ruboxistaurin and enzastaurin, which are highly active inhibitors of protein kinase C-β. While BIMs are widely recognised as protein kinase inhibitors, other modes of activity have been reported, including the inhibition of calcium signalling and antimicrobial activity. BIMs can be highly functionalised or chemically manipulated, which provides the opportunity to generate new derivatives with unique biological profiles. Critically, structural differences can be used to exploit new bioactivity and therefore it is imperative to discover new chemical entities to address new targets. This review will collate the associated bioactivities of BIM-type compounds with a focus on clinical applications
- indole
- bisindolyl
- maleimide
- BIM
- kinase inhibition
- bioactivity
- derivatives
1. Introduction
2. Bisindolylmaleimides (BIMs) and BIM-Type Inhibitors
Arcyriarubin A (BIM-IV) is the simplest bisindolylmaleimide that belongs to the family of pigments, arcyriarubin A-C (Figure 2). It was isolated from the Myxomycetes slime moulds by Steglich et al. in 1980 [9][2]. BIM-IV was a potent sub-micromolar inhibitor of protein kinase C and exhibited micromolar inhibition against seven of the other PKC isoenzymes. Fabre et al. investigated the influence of the maleimide headgroup on BIM-IV against PKC and PKA by comparison with the succinimide 79 and the lactam derivative 80 [38][3]. Both 79 and 80 were determined to have low inhibitory activity compared to BIM-IV against both PKC and PKA (Figure 2).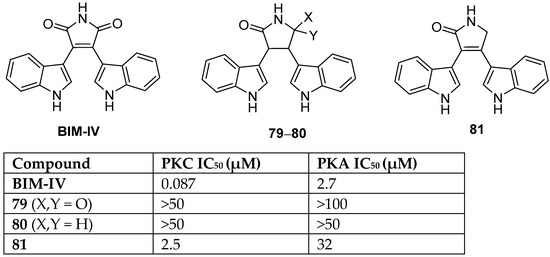
3. Selected Modified Indolylmaleimides: Benzofuranylindolylmaleimides (BfIMs)
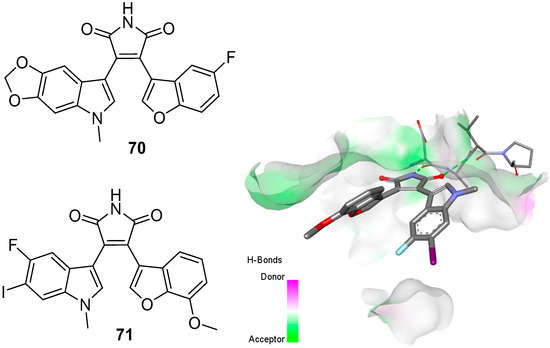
The initial incorporation of other aryl units, such as 7-azaindole, into the BIM frame proved effective in enhancing inhibitory selectivity against kinases. Benzofuran was also an attractive heterocyclic component, which was initially employed by Davis et al. to prepare the maleimide 89 [59][27]. Similar to the BIMs discussed earlier in the chapter, capping the indole nitrogen with an N-methyl group increased potency for this novel class of benzofuranylindolylmaleimides (BfIMs). Significant PKC inhibition (IC50 = 200 nM) was reported for this N-methyl compound (Figure 8).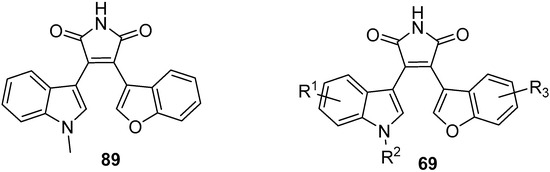
4. Selected Modified Indolylmaleimides: Napthylindolylmaleimides (NIMs)
As seen earlier, the derivatisation of the BIM frame dates back to 1992 when Davis et al. replaced one of the indole units with aryl components [59][27]. A wide panel of compounds were generated with aryl systems, including substituted phenyls, and thienyl and pyrrolyl groups. In 2006, Peifer et al. investigated arylindolyl-2,3-maleimides and evaluated their antiangiogenic activity in an in vivo assay with chick embryos. As part of this synthetic panel, naphthyl-containing maleimides were included to assess their inhibitory potential against protein kinases (Figure 10) [64][34]. All compounds were screened against twelve kinases with close attention to CDKs and PKC isoenzymes. Naphthylindolylmaleimide 73 was identified as a potent inhibitor of PKC-β1 (IC50 = 2 nM).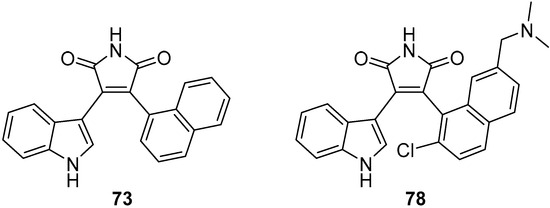
5. Recent Applications of Bisindolylmaleimides and Derivatives
In recent years, non-PKC targeted effects for BIMs have come to the fore with a number of examples of repurposing existing BIMs and the development of new BIMs to align with new targets. BIM-IX was identified by Zhang et al. to affect drug-resistant chronic myeloid leukaemia (CML) by inhibiting DNA topoisomerase and inducing cell cycle arrest and cell death [65][36]. It appears that BIM-IX is more effective than enzastaurin and other BIMs against BCR-ABL positive and T315I mutated cells and maintains its effect on in vivo cancer models through inhibition of topo IIa and B-Raf. On a similar note, both Li and Winfield et al. identified new BIM compounds with diverging activity. Li identified that the active BIM 90 bound to the SH2 domain of STAT3, and that substitution of the maleimide NH with hydroxymethyl eliminated this interaction, whereas Winfield identified that kinase inhibition could be modified by its substitution to N-OH 91. In both papers, novel N-alkylated BIMs were identified to interact with STAT3 and kinases through screening, and it is remarkable that the most active compounds contain alkyl nitrile substituents on the indole nitrogens (90 and 91, Figure 11) [28,29][37][38]. Again, moving away from PKC, Mayati et al. identified inhibition of the organic cation transporter 1 (OCT1) by BIM-IX (Ro 31-8220). This has important considerations for the activity of BIMs in drug-resistant cells and should especially be considered for other BIMs in relation to the likelihood of cellular off-target effects [66][39].
| BIM | Proposed Targets |
|---|
| BIM-Type Ligand | Kinase | Family | PDB | Reference | ||
|---|---|---|---|---|---|---|
| BIM-I | PKC, GSK-3β, 5-HT3, ABCG2, OCT-1, PDK1, MSK1, MAPKAP-K1, S6K1, Chk1, PKA, DYRK1 | |||||
| BIM-II | PKC, ABCG2, OCT-1, PDK1, MSK1, MAPKAP-K1, PKA, DYRK1 | |||||
| BIM-III | PKC, ABCG2, OCT-1, S6K1, MAPKAPK1, RSK2, MSK1, PDK1 | |||||
| BIM-IV | PKC, PKA, MAPKAPK1, MSK1, ABCG2 | |||||
| BIM-V | ABCG2, SK6 | |||||
| BIM-VI | OCT-1 | |||||
| BIM-VII | ||||||
| BIM-I | PDK1 | AGC | 1UU8 | [74][47] | ||
| OCT-1 | ||||||
| PIM1 | CAMK | 1XWS | [81][54] | |||
| PIM1 | CAMK | 2BIK | [82][55] | |||
| PIM1 | CAMK | 2BIL | [83][56 | |||
| BIM-VIII | PKC, GSK-3β, carbachol-evoked noradrenaline release, OCT-1, PDK1, MSK1, S6K1, PKA, DYRK1, MAPKAPK1 | |||||
| ] | ||||||
| PKC-1 | AGC | 1ZRZ | [84][57] | |||
| BIM-II | PDK1 | AGC | 1UU7 | [74][47] | ||
| PDK1 | AGC | 3ORZ | [85][58] | |||
| PDK1 | AGC | 3OTU | [85][58] | BIM-IX | PKC, GSK-3β, MAPKAPK1, MSK1, OCT-1, S6K1 | |
| BIM-III | PDK1 | AGC | 1UU9 | [74][47] | BIM-X | Multiple Protein Kinases |
| BIM-XI | PKC, T-cell activation |
6. X-ray Crystal Structures of Bisindolylmaleimides and Kinases
As seen in Section 3, the kinase-targeted effects of BIMs can be rationalized through crystal structure formation as ligands. To date, X-ray crystal structures have been solved for BIM compounds in a number of kinases, as detailed in Table 2. It is clear that BIM compounds are capable of forming crystal structures across the kinase families and, interestingly, the two most often quoted targets of BIMs PKC-β and GSK-3β are in the minority of solved structures. This, however, does not prevent the use of modelling to plot interactions, and indeed high throughput in silico screening has been used extensively in the last 10 years to develop the scope of targets and to rationalize the effects of these potent compounds.| BIM-VII | ||||
| DMPK1 | ||||
| AGC | ||||
| 2VD5 | ||||
| [ | 86 | ] | [ | 59] |
| BIM-VIII | PDK1 | AGC | 1UVR | [74][47] |
| Ruboxistaurin 61 | PDK1 | AGC | 1UU3 | [74][47] |
| PIM1 | CAMK | 2J2I | [87][60] | |
| BfIM 71 | GSK-3β | CMGC | 3SD0 | [88][61] |
 |
PKC-β | AGC | 2I0E | [89][62] |
 |
GSK-3β | CMGC | 2OW3 | [90][63] |
| Indolyl phenyl maleimide 94 | GSK-3β | CMGC | 1R0E | [91][64] |
| Indolyl pyrimidinyl maleimide 95 | JAK3 | TK | 3PJC | [92][65] |
| Indolyl pyrroloquinolinyl maleimide 96 | MET | TK | 3RHK | [93][66] |
| Indolyl quinazolinyl maleimide 97 | PKC-α | AGC | 3IW4 | [94][67] |
7. Conclusion
Looking at the starting BIM standards (BIM I-XI), it is clear that PKC kinase and protein kinases in general are noted targets, and so any new BIM derivative should be screened for this activity. It is also evident that multiple targets can be effectively inhibited by compounds with the bisindolylmaleimide pharmacophore and indeed selectivity within the kinases can be achieved, which suggests there is significant value in further development.
Since the early 1990s, both acyclic and macrocyclic BIM agents have excelled during in vitro studies and progressed to in vivo testing and human clinical trials. The introduction of different aryl units to the BIM pharmacophore proved to be a successful strategy to enhance target selectivity. The BIM class offers unique opportunities for drug repurposing due to its known safety profile and indeed many BIMs have completed clinical trials, which is evidence of their real-world impact. Beyond the BIM structures we have investigated, there are many other aryl maleimides [95] and bisindole derivatives [96–98] which have notable bioactivity, and hence this expanding field is sure to maintain real-world relevance in the future.
References
- Pajak, B.; Orzechowska, S.; Gajkowska, B.; Orzechowski, A. Bisindolylmaleimides in anti-cancer therapy–more than PKC inhibitors. Adv. Med. Sci. 2008, 53, 21–31.
- Steglich, W.; Steffan, B.; Kopanski, L.; Eckhardt, G. Indole Pigments from the Fruiting Bodies of the Slime Mold Arcyria denudata. Angew. Chem. 1980, 19, 459–460.
- Fabre, S.; Prudhomme, M. Protein Kinase C Inhibitors; Structure–Activity Relationships in K252c-Related Compounds. Bioorg. Med. Chem. 1993, 1, 193–196.
- Shenyin, Z.; Yulong, A. A Kind of Preparation Method of Bisindole Maleimide Compound. Chinese Patent CN105153126(A), 10 September 2015. Available online: http://patents.google.com/patent/CN105153126B/ (accessed on 9 June 2023).
- Shengyin, Z.; Zhiyu, S.; Weimin, Q.; Dengqing, Z. Research Progress of Indole Maleimide Protein Kinase C Inhibitors. Org. Chem. 2008, 28, 1676.
- Sancelme, M.; Fabre, S.; Prudhomme, M. Antimicrobial Activities of Indolocarbazole and Bis-indole Protein Kinase C Inhibitors. J. Antibiot. 1994, 47, 792–798.
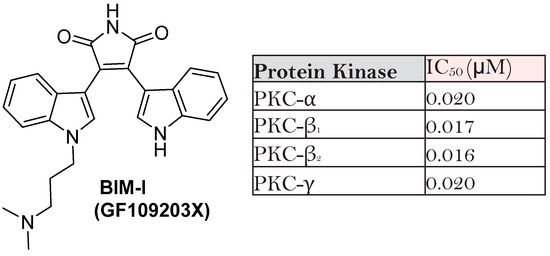
- Toullec, D.; Pianettis, P.; Coste, H.; Belleverguel, P.; Grand-Perret, T.; Ajakanee, M.; Baudet, V.; Boissin, P.; Boursier, E.; Loriolle, F.; et al. The Bisindolylmaleimide GF 109203X Is a Potent and Selective Inhibitor of Protein Kinase C. J. Biol. Chem. 1991, 266, 15771–15781.
- Hu, H. Recent discovery and development of selective protein kinase C inhibitors. Drug Discov. Today 1996, 1, 438–447.
- Zhou, F.; Huang, H.; Zhang, L. Bisindoylmaleimide I enhances osteogenic differentiation. Protein Cell 2012, 3, 311–320.
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007.
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The Protein Kinase CB–Selective Inhibitor, Enzastaurin (LY317615.HCl), Suppresses Signaling through the AKT Pathway, Induces Apoptosis, and Suppresses Growth of Human Colon Cancer and Glioblastoma Xenografts. Cancer Res. 2005, 65, 7462–7469.
- Keyes, K.A.; Mann, L.; Sherman, M.; Galbreath, E.; Schirtzinger, L.; Ballard, D.; Chen, Y.-F.; Iversen, P.; Teicher, B.A. LY317615 decreases plasma VEGF levels in human tumor xenograft-bearing mice. Cancer Chemother. Pharmacol. 2004, 53, 133–140.
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174.
- Nowakowski, G.S.; Zhu, J.; Zhang, Q.; Brody, J.; Sun, X.; Maly, J.; Song, Y.; Rizvi, S.; Song, Y.; Lansigan, F.; et al. ENGINE: A Phase III randomized placebo controlled study of enzastaurin/R-CHOP as frontline therapy in high-risk diffuse large B-cell lymphoma patients with the genomic biomarker DGM1. Future Oncol. 2020, 16, 991–999.
- A Trial of Enzastaurin Plus Temozolomide During and Following Radiation Therapy in Patients with Newly Diagnosed Glioblastoma with or without the Novel Genomic Biomarker, DGM1 2023. Available online: http://clinicaltrials.gov/study/NCT03776071 (accessed on 30 June 2023).
- Bota, D.; Butowski, N.; Piccioni, D.; De la Fuente, M.; Mao, Y.; Li, W.-B.; Trusheim, J.; Fink, K.; Campian, J.; Lobbous, M.; et al. CTNI-08. DB102-01 ENGAGE study: A biomarker-guided, randomized, double-blind, placebo-controlled, multi-center Phase 3 clinical trial of DB102 in patients with newly diagnosed glioblastoma (GBM). Neuro-Oncology 2021, 23, 60.
- Investigate Efficacy, Safety, and Pharmacokinetics of Enzastaurin for the Prevention of Arterial Events in Patients with Vascular Ehlers-Danlos Syndrome (PREVEnt). 2022. Available online: http://clinicaltrials.gov/ct2/show/NCT05463679 (accessed on 30 June 2023).
- Jirousek, M.R.; Gillig, J.R.; Gonzalez, C.M.; Heath, W.F.; McDonald, J.H.; Neel, D.A.; Rito, C.J.; Singh, U.; Stramm, L.E.; Melikian-Badalian, A.; et al. (S)-13--10,11,14,15-tetrahydro-4,9:16,21-dimetheno-1H,13H-dibenzopyrrolo oxadiazacyclohexadecene-1,3(2H)-dione (LY333531) and Related Analogues: Isozyme Selective Inhibitors of Protein Kinase Cβ. J. Med. Chem. 1996, 39, 2664–2671.
- Sobhia, M.E.; Grewal, B.K.; ML, S.P.; Patel, J.; Kaur, A.; Haokip, T.; Kokkula, A. Protein kinase C inhibitors: A patent review (2008–2009). Expert Opin. Therap. Pat. 2013, 23, 1–19.
- Teicher, B.A.; Alvarez, E.; Mendelsohn, L.G. Enzymatic rational and preclinical support for a potent protein kinase Cβ inhibitor in cancer therapy. Adv. Enzyme Regul. 1999, 39, 313–327.
- Shiying, L.; Yuyang, J.; Jian, C.; Feng, L.; Li, M.; Yufen, Z. Inhibitors of protein kinase C. Chin. Sci. Bull. 2005, 50, 1293–1304.
- McGill, J.B.; King, G.L.; Berg, P.H.; Price, K.L.; Kles, K.A.; Bastyr, E.J.; Hyslop, D.L. Clinical safety of the selective PKC-β inhibitor, ruboxistaurin. Expert Opin. Drug Saf. 2006, 5, 835–845.
- Javey, G.; Schwartz, S.G.; Flynn, H.W.; Aiello, L.P.; Sheetz, M.J. Ruboxistaurin: Review of Safety and Efficacy in the Treatment of Diabetic Retinopathy. Clin. Med. Insights Ther. 2010, 2, CMT-S5046.
- Eli Lilly Nederland, B.V. ARxxant Withdrawal Assessment Report European Medicines Agency (EMEA/H/C/000753). Available online: http://www.ema.europa.eu/en/medicines/human/withdrawn-applications/arxxant (accessed on 9 June 2023).
- Randomized and Open Label Study for Safety and Efficacy of DBI-102 vs. Vehicle vs. Hydroquinone on Skin Pigmentation and Lentigos. Available online: https://clinicaltrials.gov/study/NCT05511948 (accessed on 9 June 2023).
- Kuo, G.-H.; Prouty, C.; DeAngelis, A.; Shen, L.; O’Neill, D.J.; Shah, C.; Connolly, P.J.; Murray, W.V.; Conway, B.R.; Cheung, P.; et al. Synthesis and Discovery of Macrocyclic Polyoxygenated Bis-7-azaindolylmaleimides as a Novel Series of Potent and Highly Selective Glycogen Synthase Kinase-3β Inhibitors. J. Med. Chem. 2003, 46, 4021–4031.
- Davis, P.D.; Hill, C.H.; Lawton, G.; Nixon, J.S.; Wilkinson, S.E.; Hurst, S.A.; Keech, E.; Turner, S.E. Inhibitors of Protein Kinase, C. I. 1. 2,3-Bisarylmaleimides. J. Med. Chem. 1992, 35, 177–184.
- Kozikowski, A.P.; Gaisina, I.N.; Yuan, H.; Petukhov, P.A.; Blond, S.Y.; Fedolak, A.; Caldarone, B.; McGonigle, P. Structure-Based Design Leads to the Identification of Lithium Mimetics That Block Mania-like Effects in Rodents. Possible New GSK-3β Therapies for Bipolar Disorders. J. Am. Chem. Soc. 2007, 129, 8328–8332.
- Gaisina, I.N.; Gallier, F.; Ougolkov, A.V.; Kim, K.H.; Kurome, T.; Guo, S.; Holzle, D.; Luchini, D.N.; Blond, S.Y.; Billadeau, D.D.; et al. From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3 β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells. J. Med. Chem. 2009, 52, 1853–1863.
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small molecule Glycogen Synthase Kinase-3 inhibitor, is active in neuroblastoma. Anticancer Drugs 2018, 29, 717–724.
- 9-ING-41 in Patients with Advanced Cancers. 2019. Available online: http://www.clinicaltrials.gov/study/NCT03678883 (accessed on 30 June 2023).
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3β Inhibition with 9-ING-41 Attenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925.
- Anraku, T.; Kuroki, H.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.; Giles, F.J.; Ugolkov, A.V.; Tomita, Y. Clinically relevant GSK-3β inhibitor 9-ING-41 is active as a single agent and in combination with other antitumour therapies in human renal cancer. Int. J. Mol. Med. 2020, 45, 315–323.
- Peifer, C.; Stoiber, T.; Unger, E.; Totzke, F.; Schächtele, C.; Marme, D.; Brenk, R.; Klebe, G.; Schollmeyer, D.; Dannhardt, G. Design, Synthesis, and Biological Evaluation of 3,4-Diarylmaleimides as Angiogenesis Inhibitors. J. Med. Chem. 2006, 49, 1271–1281.
- van Eis, M.J.; Evenou, J.; Schuler, W.; Zenke, G.; Vangrevelinghe, E.; Wagner, J.; von Matt, P. Indolyl-naphthyl-maleimides as potent and selective inhibitors of protein kinase C-α/β. Bioorg. Med. Chem. Lett. 2017, 27, 781–786.
- Zhang, X.; Jia, D.; Ao, J.; Liu, H.; Zang, Y.; Azam, M.; Habib, S.L.; Li, J.; Ruan, X.; Jia, H.; et al. Identification of Bisindolylmaleimide IX as a potential agent to treat drug-resistant BCR-ABL positive leukemia. Oncotarget 2016, 7, 69945–69960.
- Li, X.; Ma, H.; Li, L.; Chen, Y.; Sun, X.; Dong, Z.; Liu, J.-Y.; Zhu, W.; Zhang, J.-T. Novel synthetic bisindolylmaleimide alkaloids inhibit STAT3 activation by binding to the SH2 domain and suppress breast xenograft tumor growth. Oncogene 2018, 37, 2469–2480.
- Winfield, H.J.; Cahill, M.M.; O’Shea, K.D.; Pierce, L.T.; Robert, T.; Ruchaud, S.; Bach, S.; Marchand, P.; McCarthy, F.O. Synthesis and anticancer activity of novel bisindolylhydroxymaleimide derivatives with potent GSK-3 kinase inhibition. Bioorg. Med. Chem. 2018, 26, 4209–4224.
- Mayati, A.; Bruyere, A.; Moreau, A.; Jouan, E.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein Kinase C-Independent inhibition of Organic Cation Transporter 1 activity by the bisindolylmaleimide Ro 31-8220. PLoS ONE 2015, 10, e0144667.
- Sosa-Peinado, A.; Fructuoso-García, K.; Vásquez-Bochm, L.X.; González-Andrade, M. Bisindolylmaleimides new ligands of CaM Protein. Molecules 2022, 27, 7161.
- Hers, I.; Tavare, J.M.; Denton, R.M. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999, 460, 433–436.
- Coultrap, S.J.; Sun, H.; Tenner, T.E., Jr.; Machu, T.K. Competitive antagonism of the mouse 5-hydroxytryptamine3 receptor by bisindolylmaleimide I, a “selective” protein kinase C inhibitor. J. Pharmacol. Exp. Ther. 1999, 290, 76–82.
- Robey, R.W.; Shukla, S.; Steadman, K.; Obrzut, T.; Finley, E.M.; Ambudkar, S.V.; Bates, S.E. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol. Cancer Ther. 2007, 6, 1877–1885.
- Deane, F.M.; Lin, A.J.S.; Hains, P.G.; Pilgrim, S.L.; Robinson, P.J.; McCluskey, A. FD5180, a Novel Protein Kinase Affinity Probe, and the Effect of Bead Loading on Protein Kinase Identification. ACS Omega 2017, 2, 3828–3838.
- Birchall, A.M.; Bishop, J.; Bradshaw, D.; Cline, A.; Coffey, J.; Elliott, L.H.; Gibson, V.M.; Greenham, A.; Hallam, T.J.; Harris, W.; et al. Ro 32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J. Pharmacol. Exp. Ther. 1994, 268, 922–929.
- Bit, R.A.; Davis, P.D.; Elliott, L.H.; Harris, W.; Hill, C.H.; Keech, E.; Kumar, H.; Lawton, G.; Maw, A.; Nixon, J.S.; et al. Inhibitors of protein kinase C. 3. Potent and highly selective bisindolylmaleimides by conformational restriction. J. Med. Chem. 1993, 36, 21–29.
- Komander, D.; Kular, G.S.; Schüttelkopf, A.W.; Deak, M.; Prakash, K.R.C.; Bain, J.; Elliott, M.; Garrido-Franco, M.; Kozikowski, A.P.; Alessi, D.R.; et al. Interactions of LY333531 and Other Bisindolyl Maleimide Inhibitors with PDK1. Structure 2004, 12, 215–226.
- Davies, S.P.; Reddy, H.; Caivano, M.; Chen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105.
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Jones, K.A.; Kathayat, R.S.; Azizi, S.-A.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H.; et al. Bisindolylmaleimide IX: A novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in-vitro validation assays. Methods 2021, 195, 57–71.
- Huang, C.; Feng, F.; Shi, Y.; Li, W.; Wang, Z.; Zhu, Y.; Yuan, S.; Hu, D.; Dai, J.; Jiang, Q.; et al. Protein Kinase C inhibitors reduce SARS-CoV-2 replication in cultured cells. Microbiol. Spectr. 2022, 10, e01056-22.
- Sun, X.; Li, L.; Ma, H.-G.; Sun, P.; Wang, Q.-L.; Zhang, T.-T.; Shen, Y.-M.; Zhu, W.-M.; Li, X. Bisindolylmaleimide alkaloid BMA-155Cl induces autophagy and apoptosis in human hepatocarcinoma HepG-2 cells through the NF-κB p65 pathway. Acta Pharmacol. Sin. 2017, 38, 524–538.
- Kumar, S.; Sannapureddi, R.K.R.; Todankar, C.S.; Ramanathan, R.; Biswas, A.; Sathyamoorthy, B.; Pradeepkumar, P.I. Bisindolylmaleimide ligands stabilize c-MYC G-Quadruplex DNA structure and downregulate gene expression. Biochemistry 2022, 61, 1064–1076.
- Yao, H.; Wang, J.; Chen, H.; Mei, X.; Su, Z.; Wu, J.; Lin, Z.; Ling, Q. Solvent-dependent and highly selective anion sensing and molecular logic application of bisindolylmaleimide derivatives. RSC Adv. 2017, 7, 12161–12169.
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–1XWS. Crystal Structure of the Human PIM1 Kinase Domain. Available online: https://doi.org/10.2210/pdb1xws/pdb (accessed on 18 July 2023).
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–2BIK. Human Pim1 Phosphorylated on Ser261. Available online: https://doi.org/10.2210/pdb2bik/pdb (accessed on 18 July 2023).
- Knapp, S.; Debreczeni, J.; Bullock, A.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Guo, K. PDB Entry–2BIL. The Human Protein Kinase Pim1 in Complex with Its Consensus Peptide Pimtide. Available online: https://doi.org/10.2210/pdb2bil/pdb (accessed on 18 July 2023).
- Messerschmidt, A.; Macieira, S.; Velarde, M.; Bädeker, M.; Benda, C.; Jestel, A.; Brandstetter, H.; Neuefeind, T.; Blaesse, M. Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J. Mol. Biol. 2005, 352, 918–931.
- Sadowsky, J.D.; Burlingame, M.A.; Wolan, D.W.; McClendon, C.L.; Jacobson, M.P.; Wells, J.A. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. USA 2011, 108, 6056–6061.
- Elkins, J.M.; Amos, A.; Niesen, F.H.; Pike, A.C.W.; Fedorov, O.; Knapp, S. Structure of dystrophia myotonica protein kinase. Protein Sci. 2009, 18, 782–791.
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Müller, S.; Bullock, A.N.; Schwaller, J.; Sundström, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528.
- Mesecar, A.M.; Walters, R.L. PDB Entry–3SD0. Identification of a Glycogen Synthase Kinase-3b Inhibitor that Attenuates Hyperactivity in CLOCK Mutant Mice. Available online: https://doi.org/10.2210/pdb3sd0/pdb (accessed on 18 July 2023).
- Grodsky, N.; Li, Y.; Bouzida, D.; Love, R.; Jensen, J.; Nodes, B.; Nonomiya, J.; Grant, S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry 2006, 45, 13970–13981.
- Zhang, H.-C.; Boñaga, L.V.R.; Ye, H.; Derian, C.K.; Damiano, B.P.; Maryanoff, B.E. Novel bis(indolyl)maleimide pyridinophanes that are potent, selective inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2007, 17, 2863–2868.
- Allard, J.; Nikolcheva, T.; Gong, L.; Wang, J.; Dunten, P.; Avnur, Z.; Waters, R.; Sun, Q.; Skinner, B. PDB Entry–1R0E. Glycogen Synthase Kinase-3 Beta in Complex with 3-Indolyl-4-Arylmaleimide Inhibitor. Available online: https://doi.org/10.2210/pdb1r0e/pdb (accessed on 18 July 2023).
- Thoma, G.; Nuninger, F.; Falchetto, R.; Hermes, E.; Tavares, G.A.; Vangrevelinghe, E.; Zerwes, H.-G. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. J. Med. Chem. 2011, 54, 284–288.
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C.K. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676.
- Wagner, J.; von Matt, P.; Sedrani, R.; Albert, R.; Cooke, N.; Ehrhardt, C.; Geiser, M.; Rummel, G.; Stark, W.; Strauss, A.; et al. Discovery of 3-(1H-indol-3-yl)-4-pyrrole-2,5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J. Med. Chem. 2009, 52, 6193–6196.