Elevated plasma lipoprotein(a) [Lp(a)] is a relatively common and highly heritable trait conferring individuals time-dependent risk of developing atherosclerotic cardiovascular disease (CVD).
- lipoprotein(a)
- hypertension
- hypertensive organ damage
- cardiovascular disease
1. Introduction
2. Lipoprotein(a) Structure, Genetics, Metabolism, Distribution of Plasma Concentration Levels, and Measurement Methods
2.1. Lp(a) Structure
Lp(a) consists of an LDL-like core lipoprotein molecule, containing apolipoprotein B (apo-B), to which a glycoprotein of variable molecular weight, apolipoprotein (a) [apo(a)], is covalently linked via a single cysteine–cysteine disulfide bond [69][57] (Figure 1). Lp(a) particles contain apo(a) and apo-B100 in a 1-to-1 molar ratio [70][58]. The lipoprotein moiety is essentially indistinguishable from LDL regarding its physical chemical properties and consists of a hydrophobic core of esterified cholesterol and triglicerides, surrounded by a surface monolayer of phospholipids, unesterified cholesterol, and other proteins [71][59]. The peculiar characteristics and the size variability of Lp(a) that is the main determinant of its plasma concentration are almost entirely accounted for by the presence of apo(a). Apo(a) is encoded by the LPA gene, located on chromosome 6q26 [72][60], and cloning of a c-DNA encoding apo(a) revealed a high degree of homology of this lipoprotein with the fibrinolytic proenzyme plasminogen [73][61]. Both molecules contain coding sequences forming multiple triple-loop structures called kringles (K) [74][62] that, due to resemblance of shape, were named after Danish pastries [75][63]. The characteristic tri-looped arrangement of the kringle structure is stabilized by the presence of three internal disulfide bridges, resulting from the interaction of six conserved cysteine residues [74][62]. Plasminogen contains an N-terminal tail domain that is attached to one copy each of five kringles, designated as kringle-1 through kringle-5, and a trypsin-like protease domain [76][64]. In contrast to plasminogen, apo(a) lacks the tail domain and the first three kringle domains of plasminogen and instead is formed of multiple repeated copies of sequences homologous to plasminogen kringle-4 (K4) domain, followed by a single kringle-5-like domain and an inactive protease-like domain [77][65]. Lp(a) contains 10 subtypes of K4 repeats (K4 type-1 to K4 type-10) that differ from each other based on aminoacidic sequence. All K4 kringle types are present in a single copy within the Lp(a) moiety, with the notable exception of K4 type-2, which is present in a variable number of identically repeated copies, usually ranging from 3 to more than 40 [70][58], that are encoded by the LPA gene. This important variation leads to the size heterogeneity in apo(a) isoforms found in the general population. As a rule, apo(a) isoform size is inversely related to plasma Lp(a) concentration in most populations [78][66]. Kringles are ligand-binding sites and as such serve critical functions and pathobiological roles that are mediated by their lysine-binding sites (LBS). K4 type-9 forms a covalent disulfide bridge to the apo-B100 moiety of LDL and is critical in the creation of the covalent apo(a) LDL-complex whose formation is crucially initiated by noncovalent interaction between LBS of apo(a) and lysine residues of apoB100. The lysine binding site in K4 type-10 is thought to mediate the binding of Lp(a) to different substrates including fibrin, cell surface receptors, and extracellular matrix proteins [79,80,81,82][67][68][69][70].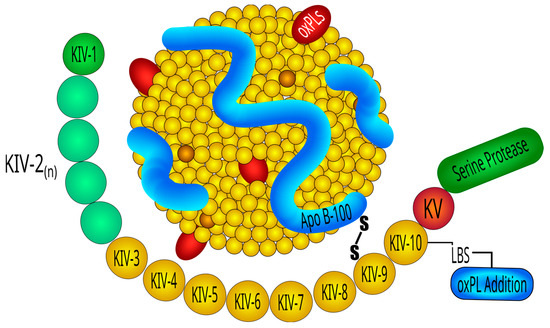
2.2. Lp(a) Genetics
2.3. Lp(a) Metabolism
Lp(a) biosynthesis faces four main steps: transcription of LPA, protein translation, transfer to the secretory pathway, and assembly of the Lp(a) particles. Lp(a) is exclusively produced in the liver which secretes apo(a)- and apoB-containing lipoproteins separately, so that the final assembly of Lp(a) takes place extracellularly by covalent linkage of apo(a) with apoB [123][87]. Synthesis and secretion are regulated by the effects of genetic control of LPA expression and processing of the apo(a) protein, respectively. Catabolism of Lp(a) is not entirely clear. Regulation and function of the endocytic receptor which removes Lp(a) from the circulation is still a matter of debate [125][88]. The presence of apo(a) and perhaps also involvement of proprotein convertase subtilisin/kexin type 9 (PCSK9) limits removal of Lp(a) by the LDL-receptor. In addition, several other endocytic receptors have been implicated to mediate removal of Lp(a) from blood, including LDL-receptor related protein 1, very low density lipoprotein receptor, scavenger receptor B1, and plasminogen receptor KT (PlgRKT) [125,126][88][89]. While most lipoprotein receptors direct Lp(a) into a route which leads to the lysosomal degradation of the entire particle, PlgRKT was reported to shuttle Lp(a) into a pathway which leads to the selective degradation of the lipids and apoB, but to the re-secretion of apo(a) which then associates with another LDL-particle to form a new Lp(a) particle.2.4. Distribution of Lp(a) Concentration and Effects of Non-Genetic Factors
The distribution of plasma Lp(a) levels is highly variable among different ethnic groups with concentrations varying up to 1000-fold within each population, ranging from less than 0.1 mg/dL to as high as 387 mg/dL. The lowest levels are seen in non-Hispanic Caucasians, Chinese, and Japanese; slightly higher levels have been documented in Hispanics, while the highest levels are found in Blacks [127][90]. In Caucasians, plasma levels are comparable in men and women, and it is estimated that 20% of the population worldwide has an Lp(a) level >50 mg/dL (>105 nmol/L) [128][91], 5% of individuals has an Lp(a) level above 120 mg/dL (250 nmol/L), whereas only 1% of individuals has an extremely elevated Lp(a) level above the 99th percentile, corresponding approximately to 180 mg/dL. Plasma levels are generally unaffected by dietary interventions or various physiological and environmental factors, including age, sex, fasting state, or physical activity, but are also known to be slightly influenced by pregnancy, menopause, hormone use, cholestasis, thyroid dysfunction, acute phase events, and renal function [129][92].
2.5. Lp(a) Measurement
Reproducible and reliable measurement of Lp(a) was difficult to obtain mainly because of the highly polymorphic nature of the apo(a) moiety, due to the variation in isoform size. Additional factors included lack of standardization across laboratories with some assays reporting Lp(a) values as mass concentrations (mg/dL) and others as particle concentrations (nmol/L) [131][93], and the adoption of antibody-based approaches which led to possible underestimation of the small isoforms and overestimation of the large isoforms [55]. The efforts of The International Federation of Clinical Chemistry and Laboratory Medicine to standardize reference material to calibrate Lp(a) assays improved the reproducibility between methods [132][94], although some degree of heterogeneity might persist [133][95].3. Plasma Lp(a) Concentrations in Hypertension
Dyslipidemia is more prevalent in hypertensive than normotensive individuals, and changes in lipid levels progressively worsen with increasing BP [135][96]. Increased levels of total and low-density lipoprotein cholesterol and triglycerides, and lower high-density lipoproteins cholesterol were reported in hypertensive patients [136][97]. Regarding Lp(a), data are highly controversial mostly depending upon lack of standardization of assays and relevant differences among ethnic groups. While some studies reported higher Lp(a) concentrations in hypertensive than normotensive subjects, other studies did not [65,137,138,139,140,141,142,143,144,145,146][98][99][100][101][102][103][104][105][106][107][108]. In a study conducted by Lip et al. on ambulatory hypertensive patients, median Lp(a) levels were found to be markedly elevated in Blacks, in line with previous observations [144][106]. Elevated plasma Lp(a) was also more frequent in hypertensive patients of Indian than Caucasian descent, although in hypertensive Caucasians no differences were observed with the respective normotensive subjects. In agreement with these findings, two additional studies reported an increased prevalence of elevated Lp(a) in Indian hypertensives free of cardiovascular complications in comparison to their respective normotensive controls [147,148][109][110].4. Lp(a) and The Vascular Wall
Essential hypertension is the most frequent form of hypertension and is characterized by a complex and multifactorial pathophysiology, where blood vessels, heart, and kidneys are reciprocally involved in regulation of the leading determinants of systemic BP, namely cardiac output and peripheral vascular resistance [149][111]. Within this complex interplay, a crucial role belongs to vascular endothelium that, in normal conditions, balances vasoconstriction and vasodilation of resistance vessels, also exerting important antithrombotic and anti-inflammatory functions that might be impaired in the process of atherogenesis [150][112]. In vitro studies indicate that elevated Lp(a) can directly contribute to atherogenesis and cause endothelial cell (ECs) and vascular smooth muscle cell (VSMCs) dysfunction. These effects appear to be prevalently mediated by the apo(a) moiety, due to its hydrophilic properties which allow a direct interaction with the vascular endothelium as well as other cellular receptors [154][113]. Much like other lipoproteins, Lp(a) can diffuse passively through endothelial surfaces via concentration gradients, accumulating in subendothelial spaces where, after binding to proteoglycans and other subendothelial structures, becomes oxidized, promotes inflammation, and mediates atherogenesis [155][114]. Lp(a) accumulation and retention in the vessel wall and sub-endothelial surfaces is facilitated by a potent lysine-binding pocket present on K4 type-10 that binds to exposed lysine on denuded endothelial surfaces and to components of the subendothelial matrix [156][115]. In animal models, high Lp(a) levels impair endothelium-dependent vasodilation, as demonstrated by a dose-dependent reduction in the expression of inducible nitric oxide synthase, both at mRNA and protein level [167,168][116][117]. Lp(a) can also affect vascular smooth muscle cells (VSMCs), as suggested by early in vitro studies showing that cell migration and proliferation could be triggered by apo(a)-mediated downregulation of plasmin-dependent activation of transforming growth factor-β [169][118].5. Lp(a) and Organ Damage in Hypertension
The development of organ damage in essential hypertensive patients is likely related, but not limited to, the direct effects of increased BP levels. Additional factors, including circulating lipids, play a crucial role in the process of vascular damage since dyslipidemia, which is frequently detected in hypertension, and serum levels of specific lipoproteins greatly affect cardiovascular morbidity and mortality in hypertensive populations [179][119]. Retrospective and prospective studies have shown that high serum levels of Lp(a) are an independent risk factor for cardiovascular diseases [180][120]. Arterial vessels are one of the main targets of hypertension and a broad interest has gathered around the mechanisms that might contribute to arterial stiffening. Arterial stiffness is recognized as a strong predictor of cardiovascular events, both in the general population [185,186][121][122] and in hypertensive patients [187,188][123][124]. Currently, arterial stiffness can be estimated by noninvasive methods, including measurements of the augmentation index (AIx) by pulse wave analysis and carotid–femoral pulse wave velocity (PWV), which provide a comprehensive assessment of the stiffness of the entire arterial tree (elastic plus muscular arteries and arterioles) [189][125]. These are now widely accepted tools for the assessment of subclinical arterial damage in hypertension [61][126]. Increased arterial stiffness has been associated with major cardiovascular risk factors, including being overweight or obese, impaired glucose tolerance, dyslipidemia, and smoking [190,191,192,193][127][128][129][130] and is characterized by both structural and functional changes of the vascular wall [194][131]. The hypothesis of a possible contribution of Lp(a) to arterial stiffening was initially investigated in elderly Japanese subjects with type 2 diabetes, reporting an independent association of its levels with the PWV [198][132]. A significant correlation between the plasma levels of oxidized Lp(a) and PWV was also reported in a subset of relatively old (mean age: 66 years) hypertensive patients with coronary artery disease and diabetes [199][133]. Similarly, in a small study enrolling hypertensive women, a significant relationship was reported between oxidized Lp(a) levels and the cardio–ankle vascular index [200][134]. All these findings might have considerable clinical implications for the identification of target organ damage in hypertensive patients as well as for their prevention and treatment. Lp(a) measurement might indeed be useful in the diagnostic workup of these patients, to identify those who might be more prone to developing organ damage. On the other hand, in consideration of the encouraging data coming from clinical trials of new Lp(a)-lowering therapies, in the near future, a reduction in Lp(a) levels might possibly improve hypertension outcomes.6. Lp(a) and Hypertensive Renal Damage
Despite robust evidence suggesting an inverse relationship between renal function and plasma Lp(a) levels in patients with severely impaired glomerular filtration rate [16[16][135],201], only a few studies have investigated this relationship in patients with hypertensive nephrosclerosis. Data obtained from large cohorts of subjects with end-stage renal disease that included mostly patients with diabetic nephropathy suggested a reciprocal relationship between Lp(a) levels and renal function. In a cross-sectional study of 417 hypertensive patients, 160 of whom had glomerular filtration rate from 30 to 89 mL/min/1.73 m2 of body surface area, scholars measured serum lipids and apolipoproteins [130][136]. Lp(a) levels were significantly higher in patients with early impairment of renal function caused by hypertensive nephrosclerosis and, most importantly, there was a highly significant inverse relationship between Lp(a) levels and glomerular filtration rate.7. Lipoprotein(a): Dietary and Pharmacological Interventions
Regarding lifestyle interventions, there is a historical assumption according to which diet has no effect on Lp(a). However, since the first report of dietary effects on Lp(a) in 1991 [210][137], there have been only a few well controlled clinical studies investigating the consequences of dietary modification on its levels. Overall, current evidence, albeit limited, indicates that diet modulates only modestly Lp(a) and often in the opposite direction to LDL-C [211,212,213,214,215,216,217,218,219][138][139][140][141][142][143][144][145][146]. The resurgence of Lp(a) in the context of cardiovascular prevention and treatment is mainly due to the relatively recent development of new RNA-directed treatments. Indeed, traditional lipid-lowering agents had demonstrated little and clinically irrelevant effects on Lp(a). These agents included statins; niacin (20% reduction of Lp(a) at maximal dose but with an unknown effectiveness in reducing major atherosclerotic cardiovascular events-MACE) [221][147]; cholesteryl ester transfer protein (20–30% reduction of Lp(a) with no reduction in MACE) [222][148]; and apolipoprotein B100 antisense oligonucleotide mipomersen (20–40% reduction in Lp(a) levels but MACE reduction unknown in patients with elevated Lp(a) with significant concerns regarding potential hepatotoxicity which justifies limited approval for homozygous familiar hypercholesterolemia) [223][149].8. Conclusions
The relationship between elevated Lp(a) levels and essential hypertension is still a matter of intriguing debate due to limited clinical evidence in support of a causal and/or reciprocal association. Nevertheless, solid evidence indicates that elevated Lp(a) levels can significantly contribute to cardiovascular and renal damage in hypertensive patients, leading to a worse clinical outcome. These effects of Lp(a) could be ascribed to the multiple detrimental effects that Lp(a) exerts on the vascular wall. Recent evidence of a longitudinal relationship of Lp(a) levels with the cardiovascular outcome in a large multiethnic cohort of hypertensive patients reinforces the need for more extensive clinical research in this field. This is further encouraged by the very promising results of the studies that have employed novel RNA-targeted treatments for Lp(a) reduction. If their benefits will be confirmed by the ongoing outcome trials, these new treatments will shift the gear for effective intervention on the residual cardiovascular risk of patients with high BP.References
- Berg, K. A new serum type system in man-the Lp(a) system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382.
- Dahlen, G.H.; Ericson, C.; Furberg, C. Myocardial infarction and an extra pre-beta lipoprotein fraction. Acta Medica Scand. 1972, 531, 25–31.
- Walton, K.W.; Hithens, J.; Magnani, H.N.; Khan, M. A study of methods of identification and estimation of Lp(a) lipoprotein and of its significance in health, hyperlipidaemia and atherosclerosis. Atherosclerosis 1974, 20, 323.
- Kostner, G.M.; Avogaro, P.; Cazzolato, G.; Marth, E.; Bittolo-Bon, G.; Qunici, G.B. Lipoprotein Lp(a) and the risk for myocardial infarction. Atherosclerosis 1981, 38, 51–61.
- Armstrong, V.W.; Cremer, P.; Eberle, E.; Manke, A.; Shulze, F.; Wieland, H.; Kreuzer, H.; Seidel, D. The association between serum Lp(a) concentrations and angiographically assessed coronary atherosclerosis. Atherosclerosis 1986, 62, 249–257.
- Rhoads, G.G.; Dahlen, G.; Berg, K.; Morton, N.E.; Dannenberg, A.L. Lp(a) lipoprotein as a risk factor for myocardial infarction. JAMA 1986, 256, 2540–2544.
- Durrington, P.N.; Ishola, M.; Hunt, L.; Arrol, S.; Bhatnagar, D. Apolipoproteins (a), AI and B and parental history in men with early onset ischaemic heart disease. Lancet 1988, 8594, 1070–1073.
- Hoefler, G.; Harnoncourt, F.; Paschke, E.; Mirtl, W.; Pfeiffer, K.H.; Kostner, G.M. Lipoprotein Lp(a): A risk factor for myocardial infarction. Arteriosclerosis 1988, 8, 398–401.
- Mbewu, A.D.; Durrington, P.N. Lipoprotein (a): Structure, properties and possible involvement in thrombogenesis and atherogenesis. Atherosclerosis 1990, 85, 1–14.
- Genest, J., Jr.; McNamara, J.R.; Ordovas, J.M.; Jenner, J.L.; Silberman, S.R.; Anderson, K.M.; Wilson, P.W.; Salem, D.N.; Schaefer, E.J. Lipoprotein cholesterol, apolipoprotein A-I and B and lipoprotein (a) abnormalities in men with premature coronary artery disease. J. Am. Coll. Cardiol. 1992, 19, 792–802.
- Abe, A.; Noma, A.; Lee, Y.J.; Yamaguchi, H. Studies on apolipoprotein (a) phenotypes Part 2 Phenotype frequen-cies and Lp(a) concentrations in different phenotypes in patients with anglographically defined coronary artery dis-ease. Atherosclerosis 1992, 96, 9–15.
- Labeur, C.; De Bacquer, D.; De Backer, G.; Vincke, J.; Muyldermans, L.; Vandekerckhove, Y.; Van der Stichele, E.; Rosseneu, M. Plasma lipoprotein (a) values and severity of eoronary artery disease in a large population of patients undergoing coronary angiography. Clin. Chem. 1992, 38, 2261–2266.
- Rosengren, A.; Wilhelmsen, L.; Eriksson, E.; Wedel, H. Lipoprotein (a) and coronary heart disease: A prospective case-control study in a general population sample of middle-aged men. BMJ 1990, 301, 1248–1251.
- Jauhiainen, M.; Koskinen, P.; Ehnholm, C.; Frick, H.M.; Manttari, M.; Manninen, V.; Huttunen, J.K. Lipoprotein(a) and coronary heart disease risk: A nested case-control study of the Helsinki Heart Study participants. Atherosclerosis 1991, 85, 59–67.
- Sigurdsson, G.; Baldursdottir, A.; Sigvaldason, H.; Agnarsson, U.; Thorgeirsson, G.; Sigfusson, N. Predictive value of apolipoproteins in a prospective survey of coronary artery disease in men. Am. J. Cardiol. 1992, 69, 1251–1254.
- Cressman, M.D.; Heyka, R.J.; Paganini, E.P.; O’Neil, J.; Skibinski, C.I.; Hoff, H.F. Lipoprotein(a) is an independent risk factor for cardiovascular disease in hemodialysis patients. Circulation 1992, 86, 475–482.
- Alfthan, G.; Pekkanen, J.; Jauhiainen, M.; Pitkdniemi, J.; Karvonen, M.; Tuomilehto, J.; Salonen, J.T.; Ehnholm, C. Relation of serum homocysteine and lipoprotein(a) concentrations to atherosclerotic disease in a prospective Finnish population based study. Atherosclerosis 1994, 106, 9–19.
- Wald, N.J.; Law, M.; Watt, H.C.; Wu, T.; Bailey, A.; Johnson, A.M.; Craig, W.Y.; Ledue, T.B.; Haddow, J.E. Apolipoproteins and ischaemic heart disease: Implications for screening. Lancet 1994, 343, 75–79.
- Bostom, A.G.; Gagnon, D.R.; Cupples, L.A.; Wilson, P.W.; Jenner, J.L.; Ordovas, J.M.; Schaefer, E.J.; Castelli, W.P. A prospective investigation of elevated lipoprotein (a) detected by electrophoresis and cardiovascular disease in women. The Framingham Heart Study. Circulation 1994, 90, 1688–1695.
- Cremer, P.; Nagel, D.; Labrot, B.; Mann, H.; Muche, R.; Elster, H.; Seidel, D. Lipoprotein Lp(a) as a predictor of myocardial infarction in comparison to fibrinogen, LDL cholesterol and other risk factors: Results from the prospective Gottingen Risk Incidence and Preva-lence Study (GRIPS). Eur. J. Clin. Investig. 1994, 24, 444–453.
- Schaefer, E.J.; Lamon-Fava, S.; Jenner, J.L.; McNamara, J.R.; Ordovas, J.M.; Davis, C.E.; Abolafia, J.M.; Lippel, K.; Levy, R.I. Lipoprotein (a) levels and risk of coronary heart disease in men. JAMA 1994, 27, 999–1003.
- Haffner, S.M.; Moss, S.E.; Klein, B.E.K.; Klein, R. Lack of association between lipoprotein (a) concentrations and coronary heart disease mortality in diabetes: The Wisconsin Epidemiologic Study of Diabetic Retinopathy. Metabolism 1992, 41, 194–197.
- Winocour, P.H.; Durrington, P.N.; Bhatnagar, D.; Mbewu, A.D.; Ishola, M.; Mackness, M.; Arrol, S. A cross-sectional evaluation of cardiovascular risk factors in coronary heart diseae associated with type 1 (insulin-dependent) diabetes mellitus. Diabetes Res. Clin. Pract. 1992, 18, 173–184.
- Ridker, P.M.; Hennekens, C.H.; Stampfer, M.J. A prospective study of lipoprotein (a) and the risk of myocardial infarction. JAMA 1993, 270, 2195–2199.
- Craig, W.Y.; Neveux, L.M.; Palomaki, G.E.; Cleveland, M.M.; Haddow, J.E. Lipoprotein(a) as a risk factor for ischemic heart disease: Metaanalysis of prospective studies. Clin. Chem. 1998, 44, 2301–2306.
- Danesh, J.; Collins, R.; Peto, R.; Lipoprotein(a) and coronary heart disease. Meta-analysis of prospective studies. Circulation 2000, 102, 1082–1085.
- Emerging Risk Factors Collaboration. Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423.
- Nave, A.H.; Lange, K.S.; Leonards, C.O.; Siegerink, B.; Doehner, W.; Landmesser, U.; Steinhagen-Thiessen, E.; Endres, M.; Ebinger, M. Lipoprotein (a) as a risk factor for ischemic stroke: A meta-analysis. Atherosclerosis 2015, 242, 496–503.
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167.
- Sultan, S.M.; Schupf, N.; Dowling, M.M.; Deveber, G.A.; Kirton Elkind, M.S. Review of lipid and lipoprotein(a) abnormalities in childhood arterial ischemic stroke. Int. J. Stroke 2014, 9, 79–87.
- Pare, G.; Caku, A.; McQueen, M.; Anand, D.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) levels and the risk of myocardial infarction among 7 ethnic groups. Circulation 2019, 139, 1472–1482.
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184.
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009, 301, 2331–2339.
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014, 63, 470–477.
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741.
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail. 2016, 4, 78–87.
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770.
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. High lipoprotein(a) and increased risk of ischemic stroke in a large contemporary general population study. J. Am. Coll. Cardiol. 2019, 74, 54–66.
- Steffen, B.; Duprez, D.; Bertoni, A.; Guan, W.; Tsai, M. Lp(a) -related risk of heart failure is evident in whites but not in other racial/ethnic groups. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2492–2504.
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22.
- Smith, G.D.; Ebrahim, S.; Lewis, S.; Hansell, A.L.; Palmer, L.J.; Burton, P.R. Genetic epidemiology and public health: Hope, hype, and future prospects. Lancet 2005, 366, 1484–1498.
- Benn, M.; Nordestgaard, B.G. From genome-wide association studies to mendelian randomization: Novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc. Res. 2018, 114, 1192–1208.
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528.
- Tregouet, D.A.; Konig, I.R.; Erdmann, J.; Munteanu, A.; Braund, P.S.; Hall, A.S.; Grosshennig, A.; Linsel-Nitschke, P.; Perret, C.; DeSuremain, M.; et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat. Genet. 2009, 41, 283–285.
- Schunkert, H.; Konig, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.R.; Barbalic, M.; Gieger, C.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338.
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512.
- Helgadottir, A.; Thorleifsson, G.; Gretarsdottir, S.; Stefansson, O.A.; Tragante, V.; Thorolfsdottir, R.B.; Jonsdottir, I.; Bjornsson, T.; Steinthorsdottir, V.; Verweij, N.; et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat. Commun. 2018, 9, 987.
- Willeit, P.; Kiechl, S.; Kronenberg, F.; Witztum, J.L.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Willeit, J.; Tsimikas, S. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): Prospective 15-year outcomes in the Bruneck study. J. Am. Coll. Cardiol. 2014, 64, 851–860.
- Albers, J.J.; Slee, A.; O’Brien, K.D.; Robinson, J.G.; Kashyap, M.L.; Kwiterovich, P.O., Jr.; Xu, P.; Marcovina, S.M. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: The AIM-HIGH trial (Atherothrombosis intervention in metabolic syndrome with low HDL/high triglyceride and impact on global health outcomes). J. Am. Coll. Cardiol. 2013, 62, 1575–1579.
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER trial (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). Circulation 2014, 129, 635–642.
- Nestel, P.J.; Barnes, E.H.; Tonkin, A.M.; Simes, J.; Fournier, M.; White, H.D.; Colquhoun, D.M.; Blankenberg, S.; Sullivan, D.R. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2902–2908.
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/AphA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350.
- Stefanutti, C.; Julius, U.; Watts, G.F.; Harada-Shiba, M.; Cossu, M.; Schettler, V.J.; De Silvestro, G.; Soran, H.; Van Lennep, J.R.; Pisciotta, L.; et al. Toward an international consensus-Integrating lipoprotein apheresis and new lipid-lowering drugs. J. Clin. Lipidol. 2017, 11, 858–871.
- Anderson, T.J.; Gregoire, J.; Pearson, G.J.; Barry, A.R.; Couture, P.; Dawes, M.; Francis, G.A.; Genest, J., Jr.; Grover, S.; Gupta, M.; et al. 2016 Canadian Cardiovascular Society Guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can. J. Cardiol. 2016, 32, 1276–1282.
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392.
- Cegla, J.; Neely, R.D.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J.; Datta, D.; Capps, N.; Shoulders, C.; Qureshi, N.; et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70.
- Utermann, G.; Weber, W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS (Fed. Eur. Biochem. Soc.) 1983, 154, 357–361.
- Albers, J.J.; Kenedy, H.; Marcovìna, S.M. Evidence that Lp(a) contains one molecole of apo(a) and one molecule of apo B: Evaluation of amino acid analysis data. J. Lipid Res. 1996, 37, 192–196.
- Sommer, A.; Prenner, E.; Gorges, R.; Stutz, H.; Grillhofer, H.; Kostner, G.M.; Paltauf, F.; Hermetter, A. Organization of phosphatidylcholine and sphingomyelin in the surface monolayer of low density lipoprotein and lipoprotein(a) as determined by time-resolved fluorometry. J. Biol. Chem. 1992, 267, 24217–24222.
- Frank, S.L.; Klisak, I.; Sparkes, R.S.; Mohandas, T.; Tomlinson, J.E.; McLean, J.W.; Lawn, R.M.; Lusis, A.J. The apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity to the homologous gene for plasminogen. Hum. Genet. 1988, 79, 352–356.
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.J.; Eaton, D.L.; Chen, E.T.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330, 132–137.
- Marcovina, M.; Albers, J.J.; Gabel, B.; Koschinsky, M.L.; Gaur, V.P. Effect of the number of apolipoprotein (a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin. Chem. 1995, 41, 246–255.
- Utermann, G. The mysteries of lipoprotein(a). Science 1989, 246, 904–910.
- Forsgren, M.; Raden, B.; Israelsson, M.; Larsson, K.; Heden, L.O. Molecular cloning and characterization of a full-length cDNA clone for human plasminogen. FEBS Lett. 1987, 213, 254–260.
- Koschinsky, M.L.; Boffa, M.B.; Marcovina, S.M. Lipoprotein(a) in cardiovascular risk assessment. In Clinical Lipidology: A Companion to Braunwald’s Heart Disease; Ballantyne, C.M., Ed.; Elsevier: New York, NY, USA, 2015; pp. 109–127.
- Harpel, P.C.; Gordon, B.R.; Parker, T.S. Plasmin catalyzes binding of lipoprotein (a) to immobilized fibrinogen and fibrin. Proc. Natl. Acad. Sci. USA 1989, 86, 3847–3851.
- Hoover-Plow, J.; Huang, M. Lipoprotein(a) metabolism: Potential sites for therapeutic targets. Metabolism 2013, 62, 479–491.
- Cai, A.; Li, L.; Zhang, Y.; Mo, Y.; Mai, W.; Zhou, Y. Lipoprotein(a): A promising marker for residual cardiovascular risk assessment. Dis. Markers 2013, 35, 551–559.
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J. Lipid Res. 2013, 54, 2815–2830.
- Scipione, C.A.; Sayegh, S.E.; Romagnuolo, R.; Tsimikas, S.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Mechanistic insights into Lp(a)-induced IL-8 expression: A role for oxidized phospholipid modification of apo(a). J. Lipid Res. 2015, 56, 2273–2285.
- Damluji, A.A.; El-Maouche, D.; Alsulaimi, A.; Martin, P.; Shamburek, R.D.; Goldberg, R.B.; Baum, S.J.; de Marchen, E.J. Accelerated atherosclerosis and elevated lipoprotein (a) after liver transplantation. J. Clin. Lipidol. 2016, 10, e434–e437.
- Barbir, M.; Khaghani, A.; Kehely, A.; Tan, K.C.; Mitchell, A.; Thompson, G.R.; Yacoub, M. Normal levels of lipoproteins including lipoprotein(a) after liver- heart transplantation in a patient with homozygous familial hypercholesterolaemia. Q. J. Med. 1992, 85, 807–812.
- Lawn, R.M.; Schwartz, K.; Patthy, L. Convergent evolution of apolipoprotein(a) in primates and hedgehog. Proc. Natl. Acad. Sci. USA 1997, 94, 11992–11997.
- Haibach, C.; Kraft, H.G.; Köchl, S.; Abe, A.; Utermann, G. The number of kringle IV repeats 3–10 is invariable in the human apo(a) gene. Gene 1998, 208, 253–258.
- Erdel, M.; Hubalek, M.; Lingenhel, A.; Kofler, K.; Duba, H.C.; Utermann, G. Counting the repetitive kringle-IV repeats in the gene encoding human apolipoprotein(a) by fibre-FISH . Nat. Genet. 1999, 21, 357–358.
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483.
- Berg, K. The Lp system. Ser. Haematol. 1968, 1, 111–136.
- Kronenberg, F. Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc. Drugs Ther. 2016, 30, 87–100.
- Laschkolnig, A.; Kollerits, B.; Lamina, C.; Meisinger, C.; Rantner, B.; Stadler, M.; Peters, A.; Koenig, W.; Stöckl, A.; Dähnhardt, D.; et al. Lipoprotein (a) concentrations, apolipoprotein (a) phenotypes, and peripheral arterial disease in three independent cohorts. Cardiovasc. Res. 2014, 103, 28–36.
- Kronenberg, F. Genetic determination of lipoprotein(a) and its association with cardiovascular disease: Convenient does not always mean better. J. Intern. Med. 2014, 276, 243–247.
- Guadagno, P.A.; Summers Bellin, E.G.; Harris, W.S.; Dayspring, T.D.; M Hoefner, D.M.; Thiselton, D.L.; Stanovick, B.; Warnick, G.R.; McConnell, J.P. Validation of a lipoprotein(a) particle concentration assay by quantitative lipoprotein immunofixation electrophoresis. Clin. Chim. Acta 2015, 439, 219–224.
- Frazer, K.A.; Narla, G.; Zhang, J.L.; Rubin, E.M. The apolipoprotein(a) gene is regulated by sex hormones and acute-phase inducers in YAC transgenic mice. Nat. Genet. 1995, 9, 424–431.
- Wade, D.P.; Puckey, L.H.; Knight, B.L.; Acquati, F.; Mihalich, A.; Taramelli, R. Characterization of multiple enhancer regions upstream of the apolipoprotein(a) gene. J. Biol. Chem. 1997, 272, 30387–30399.
- Boffelli, D.; Zajchowski, D.A.; Yang, Z.; Lawn, R.M. Estrogen modulation of apolipoprotein(a) expression: Identification of a regulatory element. J. Biol. Chem. 1999, 274, 15569–15574.
- Huby, T.; Afzal, V.; Doucet, C.; Lawn, R.M.; Gong, E.L.; Chapman, M.J.; Thillet, J.; Rubin, E.M. Regulation of the expression of the apolipoprotein(a) gene: Evidence for a regulatory role of the 5′ distal apolipoprotein(a) transcription control region enhancer in yeast artificial chromosome transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1633–1639.
- Negi, S.; Singh, S.K.; Pati, N.; Handa, V.; Chauhan, R.; Pati, U. A proximal tissue-specific module and a distal negative module control apolipoprotein(a) gene transcription. Biochem. J. 2004, 379, 151–159.
- Kostner, K.M.; Kostner, G.M. Lipoprotein (a): A historical appraisal. J. Lipid Res. 2017, 58, 1–14.
- Yeang, C.; Gordts, P.L.; Tsimikas, S. Novel Lipoprotein(a) catabolism pathway via apolipoprotein(a) recycling: Adding the plasminogen receptor PlgR (KT) to the List. Circ. Res. 2017, 120, 1050–1052.
- Sharma, M.; Redpath, G.M.; Williams, M.J.A.; McCormick, S.P.A. Recycling of apolipoprotein(a) after PlgRKT-mediated endocytosis of lipoprotein(a). Circ. Res. 2017, 120, 1091–1102.
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Boren, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853.
- Tsimikas, S.; Stroes, E.S.G. The dedicated “Lp(a) clinic”: A concept whose time has arrived? Atherosclerosis 2020, 300, 1–9.
- Ellis, K.L.; Boffa, M.B.; Sahebkar, A.; Koschinsky, M.L.; Watts, G.F. The renaissance of lipoprotein(a): Brave new world for preventive cardiology? Prog. Lipid Res. 2017, 68, 57–82.
- Scipione, C.A.; Koschinsky, M.L.; Boffa, M.B. Lipoprotein(a) in clinical practice: New perspectives from basic and translational science. Crit. Rev. Clin. Lab. Sci. 2018, 55, 33–54.
- Dati, F.; Tate, J.R.; Marcovina, S.M.; Steinmetz, A.; International Federation of Clinical Chemistry and Laboratory Medicine; IFCC Working Group for Lipoprotein(a) Assay Standardization. First WHO/IFCC International Reference Reagent for Lipoprotein(a) for Immunoassay-Lp(a) SRM 2B. Clin. Chem. Lab. Med. 2004, 42, 670–676.
- Marcovina, S.M.; Albers, J.J.; Scanu, A.M.; Kennedy, H.; Giaculli, F.; Berg, K.; Couderc, R.; Dati, F.; Rifai, N.; Sakurabayashi, I.; et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin. Chem. 2000, 46, 1956–1967.
- Borghi, C. Interactions between hypercholesterolemia and hypertension: Implications for therapy. Curr. Opin. Nephrol. Hypertens. 2002, 5, 489–496.
- Gebrie, A.; Gnanasekaran, N.; Menon, M.; Sisay, M.; Zegeye, A. Evaluation of lipid profiles and hematological parameters in hypertensive patients: Laboratory-based cross-sectional study. SAGE Open Med. 2018, 6, 2050312118756663.
- Sechi, L.A.; Kronenberg, F.; De Carli, S.; Falleti, E.; Zingaro, L.; Catena, C.; Utermann, G.; Bartoli, E. Association of lipoprotein(a) levels and apolipoprotein(a) polymorphism with target-organ damage in arterial hypertension. JAMA 1997, 277, 1689–1695.
- Gazzaruso, C.; Buscaglia, P.; Garzaniti, A.; Bonetti, G.; Savino, S.; Mariotti, S.; Jucci, A.; Finardi, G.; Geroldi, D. Lipoprotein(a) plasma concentrations, apolipoprotein (a) polymorphism and family history of coronary heart disease in patients with essential hypertension. J. Cardiovasc. Risk 1996, 3, 191–197.
- Constans, J.; Wendling, G.; Peuchant, E.; Camilleri, G.; Conri, C. Lipoprotein(a) in 505 hospitalized patients with various pathological states: Correlations with cardiovascular diseases and therapies. Int. Angiol. 1996, 15, 1–5.
- Saku, K.; Liu, K.; Takeda, Y.; Jimi, S.; Arakawa, K. Effects of lisinopril and bisoprolol on lipoprotein metabolism in patients with mild-to-moderate essential hypertension. Clin. Ther. 1995, 17, 1136–1146.
- Van Wersch, J.W. The behaviour of lipoprotein(a) in patients with various diseases. Scand. J. Clin. Lab. Investig. 1994, 54, 559–562.
- Glueck, C.J.; Glueck, H.I.; Hamer, T.; Speirs, J.; Tracy, T.; Stroop, D. Beta blockers, Lp(a), hypertension, and reduced basal fibrinolytic activity. Am. J. Med. Sci. 1994, 307, 317–324.
- Zhuang, Y.Y.; Wang, J.J.; Xu, P. Increased lipoprotein (a) as an independent risk factor for cardiovascular and cerebro-vascular diseases. Chin. Med. J. 1993, 106, 597–600.
- Catalano, M.; Perilli, E.; Carzaniga, G.; Colombo, F.; Carotta, M.; Andreoni, S. Lp(a) in hypertensive patients. J. Hum. Hypertens. 1998, 12, 83–89.
- Lip, G.Y.; Blann, A.D.; Jones, A.F.; Lip, P.L.; Beevers, D.G. Relation of endothelium, thrombogenesis, and hemorheology in systemic hypertension to ethnicity and left ventricular hypertrophy. Am. J. Cardiol. 1997, 80, 1566–1571.
- Ward, N.C.; Nolde, J.M.; Chan, J.; Carnagarin, R.; Watts, G.F.; Schlaich, M.P. Lipoprotein (a) and hypertension. Curr. Hypertens. Rep. 2021, 23, 44.
- Lima, L.M.; Carvalho, M.d.G.; Loures-Vale, A.A.; Fernandes, A.P.; Mota, A.P.; Neto, C.P.; Garcia, J.C.F.; Saad, J.A.; Souza, M.D.O. Increased serum levels of lipoprotein (a) correlated with the severity of coronary artery disease in patients submitted to angiography. Arq. Bras. Cardiol. 2006, 86, 506–511.
- Mahto, S.K.; Sheoran, A.; Gadpayle, A.K.; Gupta, K.; Gupta, P.K.; Chitkara, A.; Agarwal, N. Evaluation of lipoprotein (a) and lipid abnormalities in patients with newly detected hypertension and its association with severity of hypertension. J. Fam. Med. Prim. Care 2022, 11, 1508–1513.
- Bhavani, B.A.; Padma, T.; Sastry, B.; Reddy, N.K. Plasma Lipoprotein (a) levels in patients with untreated essential hyper-tension. Indian J. Hum. Genet. 2003, 9, 65–68.
- Coffman, T.M. Under pressure: The search for the essential mechanisms of hypertension. Nat. Med. 2011, 17, 1402–1409.
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42.
- Tsimikas, S. Potential causality and emerging medical therapies for lipoprotein(a) and its associated oxidized phos-pholipids in calcific aortic valve stenosis. Circ. Res. 2019, 124, 405–415.
- Nielsen, L.B.; Grønholdt, M.L.; Schroeder, T.V.; Stender, S.; Nordestgaard, B.G. In vivo transfer of lipoprotein(a) into human atherosclerotic carotid arterial intima. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 905–911.
- Hoover-Plow, J.L.; Miles, L.A.; Fless, G.M.; Scanu, A.M.; Plow, E.F. Comparison of the lysine binding functions of lipopro-tein(a) and plasminogen. Biochemistry 1993, 32, 13681–13687.
- Moeslinger, T.; Friedl, R.; Volf, I.; Brunner, M.; Koller, E.; Spieckermann, P.G. Inhibition of inducible nitric oxide synthesis by oxidized lipoprotein(a) in a murine macrophage cell line. FEBS Lett. 2000, 478, 95–99.
- Rubanyi, G.M.; Freay, A.D.; Lawn, R.M. Endothelium-dependent vasorelaxation in the aorta of transgenic mice expressing human apolipoprotein(a) or lipoprotein(a). Endothelium 2000, 7, 253–264.
- O’Neil, C.H.; Boffa, M.B.; Hancock, M.A.; Pickering, J.G.; Koschinsky, M.L. Stimulation of vascular smooth muscle cell proliferation and migration by apolipoprotein(a) is dependent on inhibition of transforming growth factor-beta activation and on the presence of kringle IV type 9. J. Biol. Chem. 2004, 279, 55187–55195.
- Mancia, G.; Volpe, R.; Boros, S.; Ilardi, M.; Giannattasio, C. Cardiovascular risk profile and blood pressure control in Italian hypertensive patients under specialist care. J. Hypertens. 2004, 22, 51–57.
- Iannuzzo, G.; Tripaldella, M.; Mallardo, V.; Morgillo, M.; Vitelli, N.; Iannuzzi, A.; Aliberti, E.; Giallauria, F.; Tramontano, A.; Carluccio, R.; et al. Lipoprotein(a) where do we stand? from the physiopathology to innovative terapy. Biomedicines 2021, 9, 838.
- Mattace-Raso, F.U.S.; van der Cammen, T.J.M.; Hofman, A.; van Popele, N.M.; Bos, M.L.; Schalekamp, M.A.D.H.; Asmar, R.; Reneman, R.S.; Hoeks, A.P.G.; Bretler, M.M.B.; et al. Arterial stiffness and risk of coronary heart disease and stroke: The Rotterdam Study. Circulation 2006, 113, 657–663.
- Willum-Hansen, T.; Staessen, J.A.; Torp-Pedersen, C.; Rasmussen, S.; Thijs, L.; Ibsen, H.; Jeppesen, J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation 2006, 113, 664–670.
- Laurent, S.; Boutouyrie, P.; Asmar, R.; Gautier, I.; Laloux, B.; Guize, L.; Ducimetiere, P.; Benetos, A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 2001, 37, 1236–1241.
- Laurent, S.; Katsahian, S.; Fassot, C.; Tropeano, A.I.; Gautier, I.; Laloux, B.; Boutouyrie, P. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke 2003, 34, 1203–1206.
- Townsend, R.R.; Wilkinson, I.B.; Schiffrin, E.L.; Avolio, A.P.; Chirinos, J.A.; Cockroft, J.R.; Heffernan, K.S.; Lakatta, E.G.; McEniery, C.M.; Mitchell, G.F.; et al. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement From the American Heart Association. Hypertension 2015, 66, 698–722.
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104.
- Wildman, R.P.; Farhat, G.N.; Patel, A.S.; Mackey, R.H.; Brockwell, S.; Thompson, T.; Sutton-Tyrrell, K. Weight change is associated with change in arterial stiffness among healthy young adults. Hypertension 2005, 45, 187–192.
- Catena, C.; Colussi, G.; Frangipane, A.; Russo, A.; Verheyen, N.D.; Sechi, L.A. Carotid artery stiffness is related to hyperinsulinemia and insulin-resistance in middle-aged, non-diabetic hypertensive patients. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 968–974.
- Wilkinson, I.B.; Prasad, K.; Hall, I.R.; Thomas, A.; MacCallum, H.; Webb, D.J.; Frenneaux, M.P.; Cockroft, J.R. Increased central pulse pressure and augmentation index in subjects with hypercholesterolemia. J. Am. Coll. Cardiol. 2002, 39, 1005–1011.
- Jatoi, N.A.; Jerrard-Dunne, P.; Feely, J.; Mahmud, A. Impact of smoking and smoking cessation on arterial stiffness and aortic wave reflection in hypertension. Hypertension 2007, 49, 981–985.
- Luft, F.C. Molecular mechanisms of arterial stiffness: New insights. J. Am. Soc. Hypertens. 2012, 6, 436–438.
- Wakabayashi, I.; Masuda, H. Lipoprotein (a) as a determinant of arterial stiffness in elderly patients with type 2 diabetes mellitus. Clin. Chim. Acta 2006, 373, 127–131.
- Morishita, R.; Ishii, J.; Kusumi, Y.; Yamada, S.; Komai, N.; Ohishi, M.; Nomura, M.; Hishida, H.; Niihashi, M.; Mitsumata, M. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: Potential role of oxidized lipoprotein(a) in the vasucular wall. J. Atheroscl. Thromb. 2009, 16, 410–418.
- Kotani, K.; Yamada, S.; Yamada, T.; Kario, K.; Taniguchi, N. Oxidized lipoprotein(a) and cardio-ankle vascular index (CAVI) in hypertensive subjects. Heart Vessel. 2013, 28, 461–466.
- Kronenberg, F.; Kathrein, H.; König, P.; Neyer, U.; Sturm, W.; Lhotta, K.; Gröchenig, E.; Utermann, G.; Dieplinger, H. Apolipoprotein(a) phenotypes predict the risk for carotid atherosclerosis in patients with end-stage renal disease. Arterioscler. Thromb. 1994, 14, 1405–1411.
- Sechi, L.A.; Zingaro, L.; De Carli, S.; Sechi, G.; Catena, C.; Falleti, E.; Dell’Anna, E.; Bartoli, E. Increased serum lipoprotein(a) levels in patients with early renal failure. Ann. Intern. Med. 1998, 129, 457–461.
- Hornstra, G.; Van Houwelingen, A.C.; Kester, A.D.; Sundram, K. A palm oil-enriched diet lowers serum lipoprotein (a) in normocholesterolemic volunteers. Atherosclerosis 1991, 90, 91–93.
- Ginsberg, H.N.; Kris-Etherton, P.; Dennis, B.; Elmer, P.J.; Ershow, A.; Lefevre, M.; Pearson, T.; Roheim, P.; Ramakrishnan, R.; Reed, R.; et al. Effects of reducing dietary saturated fatty acids on plasma lipids and lipoproteins in healthy subjects: The DELTA Study, protocol 1. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 441–449.
- Berglund, L.; Lefevre, M.; Ginsberg, H.N.; Kris-Etherton, P.M.; Elmer, P.J.; Stewart, P.W.; Ershow, A.; Pearson, T.A.; Dennis, B.H.; Roheim, P.S.; et al. Comparison of monounsaturated fat with carbohydrates as a replacement for saturated fat in subjects with a high metabolic risk profile: Studies in the fasting and postprandial states. Am. J. Clin. Nutr. 2007, 86, 1611–1620.
- Clevidence, B.A.; Judd, J.T.; Schaefer, E.J.; Jenner, J.L.; Lichtenstein, A.H.; Muesing, R.A.; Wittes, J.; Sunkin, M.E. Plasma lipoprotein (a) levels in men and women consuming diets enriched in saturated, cis-, or trans-monounsaturated fatty acids. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1657–1661.
- Muller, H.; Lindman, A.S.; Blomfeldt, A.; Seljeflot, I.; Pedersen, J.I. A diet rich in coconut oil reduces diurnal postprandial variations in circulating tissue plasminogen activator antigen and fasting lipoprotein (a) compared with a diet rich in unsaturated fat in women. J. Nutr. 2003, 133, 3422–3427.
- Müller, H.; Lindman, A.S.; Brantsæter, A.L.; Pedersen, J.I. The serum LDL/HDL cholesterol ratio is influenced more favorably by exchanging saturated with unsaturated fat than by reducing saturated fat in the diet of women. J. Nutr. 2003, 133, 78–83.
- Faghihnia, N.; Tsimikas, S.; Miller, E.R.; Witztum, J.L.; Krauss, R.M. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J. Lipid Res. 2010, 51, 3324–3330.
- Berryman, C.E.; West, S.G.; Fleming, J.A.; Bordi, P.L.; Kris-Etherton, P.M. Effects of daily almond consumption on cardiometabolic risk and abdominal adiposity in healthy adults with elevated LDL-cholesterol: A randomized controlled trial. J. Am. Heart Assoc. 2015, 4, e000993.
- Jenkins, D.J.; Kendall, C.W.; Marchie, A.; Parker, T.L.; Connelly, P.W.; Qian, W.; Haight, J.S.; Faulkner, D.; Vidgen, E.; Lapsley, K.G. Dose response of almonds on coronary heart disease risk factors: Blood lipids, oxidized low-density lipoproteins, lipoprotein (a), homocysteine, and pulmonary nitric oxide: A randomized, controlled, crossover trial. Circulation 2002, 106, 1327–1332.
- Fraley, A.E.; Schwartz, G.G.; Olsson, A.G.; Kinlay, S.; Szarek, M.; Rifai, N.; Libby, P.; Ganz, P.; Witztum, J.L.; Tsimikas, S.; et al. Relationship of oxidized phospholipids and biomarkers of oxidized low-density lipoprotein with cardiovascular risk factors, inflammatory biomarkers, and effect of statin therapy in patients with acute coronary syndromes: Results from the MIRACL (Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering) trial. J. Am. Coll. Cardiol. 2009, 53, 2186–2196.
- Croyal, M.; Ouguerram, K.; Passard, M.; Ferchaud-Roucher, V.; Chétiveaux, M.; Billon-Crossouard, S.; de Gouville, A.C.; Lambert, G.; Krempf, M.; Nobécourt, E. Effects of extended-release nicotinic acid on apolipoprotein (a) kinetics in hypertriglyceridemic patients. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2042–2047.
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 2017, 376, 1933–1942.
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184.