Figure 5. The LAT adaptor acts as a signaling scaffold. (
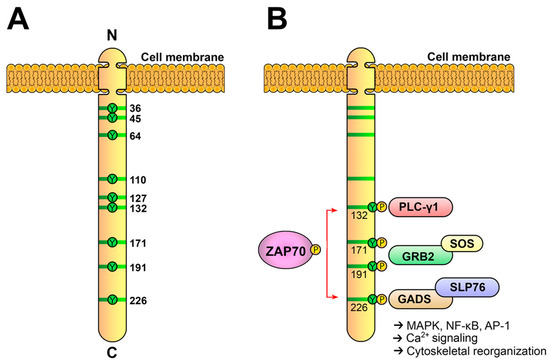
A) Schematic representation of human LAT. LAT is an integral membrane adaptor with a short N-terminal extracellular region, a transmembrane region and a long cytoplasmic tail. This tail contains nine conserved tyrosine residues that are phosphorylated to give rise to new binding sites for the SH2 domains of proteins such as PLC-γ1, Grb2 and Gads. (
B) LAT signalosome formation. The LAT signalosome is a multiprotein complex formed by LAT, PLC-γ1, Grb2, Gads and SLP-76.
The first functional analyses of LAT in cell lines demonstrated that this adaptor is essential for the generation of calcium fluxes or the activation of the transcription factor NFAT, as LAT-deficient cells showed no deficiency in proximal signaling events, but downstream pathways were not activated in these cells
[68]. Moreover, the four carboxy-terminal tyrosine residues of LAT were required for the activation of downstream signaling
[69][70][69,70]. This kind of analysis established that conserved tyrosines 7, 8 and 9 (171, 191 and 226 in human LAT) share the responsibility of binding to the SH2 domain of Grb2, tyrosine 6 of LAT (132 in human LAT), binds upon its phosphorylation to the C-terminal SH2 domain of PLC-γ1 and the Gads-SLP-76 complex binds to phosphorylated tyrosines 7 and 8
[65][69][70][65,69,70]. It has been proposed that the LAT cytoplasmic tail consists of a long flexible segment constituting a kind of “protein fishing line” encompassing several SH2- and/or PTB-binding motifs
[10]. Because of its unstructured nature, the cytoplasmic segment of LAT has a larger capture radius than a compact, folded protein with restricted conformational flexibility. After the TCR-induced phosphorylation of the four C-terminal tyrosines of LAT, the recruitment of Grb2, Gads-SLP-76 and PLC-γ1 occurs and a LAT signalosome is formed, which allows for the activation of downstream pathways (
Figure 5B).
The need for the LAT adaptor for T-cell development was formally demonstrated after the generation of a mouse
LAT-knockout strain (
Lat−/−)
[71]. The most remarkable result of LAT deficiency was the total absence of peripheral αβ and γδ T cells due to an early block of thymic development at the CD4−CD8− DN stage. Interestingly, NK cells, which also express LAT, were phenotypically and functionally normal, initially discarding any role for this adaptor in NK-cell biology
[71][72][71,72]. Next, the in vivo role of the four C-terminal tyrosine residues of LAT was analyzed by means of a knockin strain harboring Tyr-to-Phe mutations at positions 6, 7, 8 and 9 (136, 175, 195 and 235 in mouse LAT,
Lat4YF)
[73]. The phenotype of
Lat4YF knockin mice was identical to that of
Lat−/− mice: there was no peripheral T lymphocytes because of a block in T-cell development at the DN stage, but no effect was observed on B cells or NK cells. These results demonstrate that the distal four tyrosine residues of LAT are essential for pre-TCR signaling and T-cell development in vivo, suggesting that the assembly of the LAT signalosome is critical for the β-selection checkpoint at the DN stage. More support for the relevance of the four C-terminal tyrosine residues was obtained by means of adoptive transfer into irradiated
Lat−/− mice with bone-marrow cells (also from
Lat−/− mice) transduced with a retroviral vector coding for wild-type or different LAT mutants
[74]. The adoptive transfer of irradiated
Lat−/− mice with bone-marrow cells expressing a LAT mutant containing only the four distal tyrosines (i.e., having the first five tyrosines mutated to phenylalanines) restored thymic development similar to bone-marrow cells expressing wild-type LAT, indicating that the four C-terminal tyrosine residues of LAT were sufficient for T-cell development.
In this context, it was quite surprising that the phenotype of a knockin strain of mice expressing a LAT mutant in which the sixth tyrosine had been mutated to phenylalanine (
LatY136F)
[75][76][75,76]. This tyrosine residue is located in a conserved consensus sequence (YLVV) for the binding of PLC-γ1
[77], and its mutation abrogated the LAT-PLC-γ1 interaction and Ca
2+ influx generation in Jurkat cells
[69][78][69,78]. Concordantly, young
LatY136F mice showed a partial block in thymic development, with thymi that contained approximately tenfold fewer cells than their wild-type counterparts, and reduced numbers of CD4+CD8+ DP cells. Interestingly, in mice older than 7 weeks, DP cells were almost undetectable, and coincident with the loss of DP cells, a population of CD4 T cells started to dominate the thymus, which corresponded to abnormal peripheral CD4 cells that expanded in the periphery of
LatY136F mice. Indeed, spleen and lymph nodes from knockin mice were greatly enlarged and contained a population of CD4+ T cells with a phenotype (CD44
high, CD62L
low, CD69
+ and CD24
−) resembling activated/memory T cells and spontaneously producing high amounts of T
H2 type cytokines. These cytokines were responsible for the eosinophilia observed in the thymi and periphery of these mice and the observed augmented population of activated B cells producing high amounts of IgG1 and IgE. Therefore, besides its role as a transducer of activatory signals from the TCR/pre-TCR, LAT showed, for the first time, its role as a regulator of the homeostasis of the T-cell compartment.
It is also of interest to highlight the potential role of LAT in the clustering of the signaling elements of the TCR signalosome. It has been shown that LAT-Grb2-Sos1 interaction is able to generate liquid condensates at the plasma membrane, which are capable of triggering TCR signaling due to their ability to concentrate signaling reactants, as well as to exclude CD45
[79][80][82,83]. Indeed, it has been very recently shown that LAT condensates are thermodynamically coupled to ordered membrane domains during T-cell activation, demonstrating that coupling of membrane domains and cytoplasmic condensates via LAT is essential for activating signaling downstream of TCR ligation
[81][84].
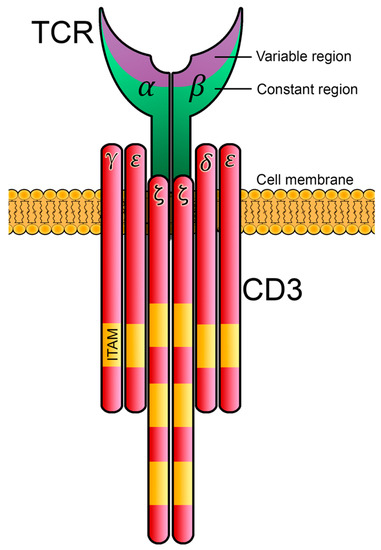
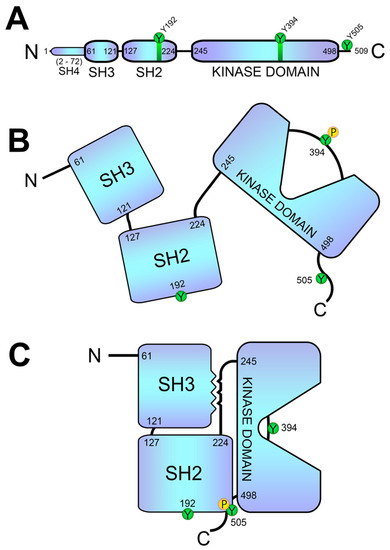
6. Regulation of Lck Activity
As previously discussed, Lck is the first kinase to act upon TCR engagement. The phosphorylation of residues Y394 and Y505 correlates with the different conformations of Lck, which have been classically related to its activity. CD45 is crucial for Y505 dephosphorylation and the opening of the Lck conformation. The CD45 phosphatase possesses a large extracellular domain that is highly glycosylated, which is required for optimal TCR signaling
[82][85]. Interestingly,
in this work, Acuto and coworkers also showed that Lck is phosphorylated at Y394 in unstimulated Jurkat cells. This
study challenged the model, in which it was TCR engagement that served to dephosphorylate Y505 and consequently open the Lck structure. More support for a model in which the opening of the Lck structure was not the only requisite for TCR signaling came from FRET analysis, which did not detect any conformational change in Lck after TCR ligation
[83][86]. Shortly thereafter, the same group demonstrated that relatively high amounts of Lck kinase phosphorylated at Y394 are expressed in resting T cells and thymocytes
[84][87]. In their work, they also showed that approximately half of the Lck molecules phosphorylated at Y394 were also phosphorylated at the inhibitory Y505 residue, constituting what was observed, for the first time, as a doubly phosphorylated Lck (DPho-Lck). Interestingly, this Dpho-Lck isoform showed the same kinase activity as Lck phosphorylated only at Y394. Moreover, in their work, Nika et al. showed that TCR stimulation did not increase the activation of Lck (measured as phosphorylation at Y394), contrary to other downstream signals. Given that the enzymatic inhibition of Lck prevented TCR signaling, these data suggested that a pool of Lck molecules are basally preactivated to phosphorylate ITAMs as soon as they are located in the proximity, but if the Y394 residue is dephosphorylated, this would block TCR signaling. Whether the Lck activity upon productive TCR engagement is increased or not is still the subject of debate. Although the maintenance of the pool of Lck phosphorylated at Y394 is necessary for the generation of TCR proximal signals, it has been shown that this active pool of Lck in resting T cells might actually be smaller than originally estimated
[85][88].
This therefore established that, in resting T cells, there was a pool of Lck molecules ready to act. The question then was whether the pool of activated molecules was fixed or depended on an equilibrium governed by the competing activities of CD45 and Csk kinase (both acting on the Y505 residue of Lck). In a fixed-pool model, the activation of Lck would require changes in its localization for the initiation of the signaling cascade, whereas in the dynamic equilibrium model, a small change in the CD45 or Csk activities would be sufficient to alter the Lck activity and initiate TCR signaling. By means of expressing a mutant form of the Csk (Csk
AS) kinase that could be inhibited very quickly and specifically, Arthur Weiss’ group showed that the inhibition of Csk
AS resulted in potent and sustained signal transduction and cell activation independent of TCR ligation
[86][89]. These data suggested that there is a continuous turnover of the Y394 and Y505 phosphorylation of Lck in resting cells, allowing for the rapid and efficient phosphorylation of ITAMs by Lck in response to TCR stimulation. Later on, the same group generated mice expressing only Csk
AS by means of BAC transgenesis into heterozygous Csk
+/− mice (as Csk knockout is lethal during embryonic development)
[87][88][90,91]. The inhibition of Csk
AS in thymocytes induced a rapid hyperactivation of Lck, which, in turn, induced the phosphorylation of proximal TCR-signaling components. However, the inhibition of Csk
AS did not increase Erk phosphorylation or intracellular Ca
2+, revealing that the initial activation of Lck was a necessary but not sufficient requirement for the full activation of the TCR-associated-signaling cascade. Moreover, using this murine model, it was shown that Csk inhibition during TCR stimulation generated more potent and prolonged intracellular signals
[89][92]. Interestingly, Csk inhibition enhanced immune responses to very weak antigens, revealing the important role of the negative regulation of Lck by Csk in establishing the signaling threshold and self/non-self-discrimination of the TCR. The model of the interplay between the opposing enzymatic activities of CD45 and Csk is not incompatible with other constraints on the initiation of TCR signaling. For example, it has been shown that the rigid extracellular domain (ECD) of CD45 sterically excludes from the membrane sites of TCR-ligand engagement
[90][93]. It is of interest to note that Csk is a cytosolic protein, and for the negative regulation of Lck activity, it has to be located at the plasma membrane. One of the candidates for recruiting Csk to the plasma membrane is the transmembrane adaptor phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG)
[91][94]. PAG is strongly tyrosine-phosphorylated in unstimulated T cells, allowing it to bind to Csk. Upon TCR stimulation, PAG is dephosphorylated and dissociates from Csk, which would diminish or abolish the suppressive effect of Csk on Lck
[91][92][94,95]. Moreover, although PAG KO mice do not exhibit obvious alterations in T-cell development or naive T-cell phenotype, effector T cells from these mice show increased activation responses
[93][96]. TCR-stimulated PAG KO effector T cells also show increased phosphorylation of ZAP70 and PLC-γ1 and increased calcium fluxes compared to wild-type T cells, although little or no increase in the phosphorylation of other downstream effectors such as Erk, Akt or JNK.
It therefore seems clear that the balance between the phosphorylated and dephosphorylated states of Y505 and Y394 of Lck, governed in turn by Csk and CD45, establishes an activation threshold for the TCR-signaling cascade. This raises the question, however, of how the recruitment of Csk (a cytosolic enzyme) to the plasma membrane is regulated. Recent evidence has shown that the PD-1 ligation induces PAG phosphorylation and, thus, Csk recruitment
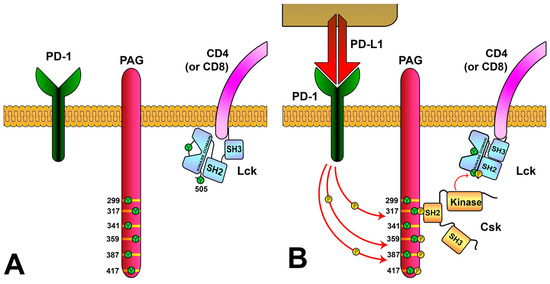
[94][97] (
Figure 6). Interestingly, this study also shows a correlation of the PAG expression levels with the increased survival from several tumor types. This study, apart from suggesting that PAG is a critical mediator of PD-1 signaling and thus a potential target for enhancing the anti-tumor action of CAR-T cells, proposes a mechanism by which PD-1, and perhaps other membrane molecules, participate in the negative-feedback loops of the TCR-signaling cascade.
Figure 6. Regulation of Lck activity via the PD-1/PAG pathway. (
A) PAG is located in the T-cell membrane and has multiple tyrosine residues in their cytoplasmic region. (
B) PD-L1 binding to PD-1 induces the phosphorylation of PAG tyrosines, generating binding sites for Csk. The SH2 domain of Csk binds to PAG-phosphorylated tyrosine 317, causing a conformational change that increases its kinase activity. Csk localizes close to the membrane and phosphorylates tyrosine 505 of Lck, inducing Lck to its inactive conformation.
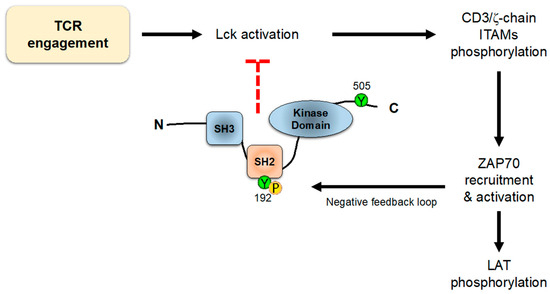
On the other hand, it has been shown that residues Y394 and Y505 are not the only ones that regulate the conformation and/or activity of Lck. In an interesting work, Weiss and colleagues showed an unpredicted role for a tyrosine residue (Y192) within the SH2 domain of Lck
[95][98]. Mutation of this residue strikingly decreases TCR early signaling, including ZAP70, LAT, PLC-γ1 and Erk phosphorylation, and calcium influx generation. These data suggest that Y192 of Lck allows for the interaction with CD45, which in turn is able to dephosphorylate Y505 and stabilize the open conformation of Lck. Therefore, TCR engagement induces Lck-mediated ZAP70 activation, which in turn triggers signaling events, also including negative-feedback loops; one of these loops consists in the phosphorylation of tyrosine 192 of Lck, which disrupts the ability of CD45 to interact with and activate Lck via the dephosphorylation of Y505 (
Figure 7). Interestingly, the defects observed in TCR-associated signals produced by mutations in Y192 are strikingly similar to the phenotype originated by CD45 loss, which supports the role of Lck Tyr192 in the interaction with this phosphatase. Accordingly, cells expressing Lck-Y192A or Lck-Y192E mutants show the increased phosphorylation of this inhibitory C-terminal tyrosine residue
[95][98]. This is an unusual regulatory mechanism by which a phosphorylation event triggers the uncoupling of two proteins, and this should be the reason why the mutation to phenylalanine of tyrosine 192 does not have a negative impact on TCR signaling: Phe residues could be structurally not so different from Tyr residues, and are still able to bind to CD45, while Tyr to Ala or Glu induces a strong modification of the tertiary structure of the SH2 domain, preventing binding to CD45 and thus leading to a high basal level of Tyr505 phosphorylation, which sharply reduces the pool of active Lck. Accordingly, knockin mice expressing a Tyr to Glu (Y192E) form of Lck showed a strong decrease in the total thymocyte numbers, with reduced numbers of DP and SP populations and defective positive and negative selection
[96][97][99,100]. Interestingly, T cells from LckY192E knockin mice showed a diminished binding to CD45 and a concomitant hyperphosphorylation of Y505, thus corroborating previous data obtained from Jurkat T cells. Surprisingly however, KI mice with a Tyr-to-Phe mutation (Y192F), which prevents its phosphorylation by ZAP70, do not appear to show any alteration in thymic development
[96][99]. If Y192 phosphorylation is part of a negative-feedback loop, then an increase in T-cell activation, or alterations in thymic development, would be expected. More work will be needed to clarify these points.
Figure 7. Lck negative-feedback loop. TCR engagement activates intracellular signaling leading to ITAMs phosphorylation and ZAP70 recruitment. The activation of ZAP70 leads not only to downstream activation signaling, but also to the phosphorylation of Y192 of Lck, which inhibits its binding to CD45 phosphatase, allowing the phosphorylation of Y505 and promoting the closed Lck conformation.
Regarding the functional regulation of Lck, it should be noted that, as a membrane protein, its interaction with membrane lipids affects its localization and functions. Lipids surrounding integral membrane proteins (termed boundary lipids or lipid sheaths) can contribute to the structure and function of these proteins. Two hypotheses exist to explain the interactions between integral membrane proteins. First, the raft hypothesis postulates that, below a critical temperature, there are relatively stable membrane domains of about 100 nm in which specific lipids and proteins assemble. A second model stipulates that more diffuse lipid- and protein-density fluctuations occur in the plasma membrane that are sufficient to promote some molecular encounters, while making others less likely, consequently leading to membrane organization. Although there is evidence to support a general biophysical mechanism for the reorganization of the local order of membranes
[98][99][100][101,102,103], recently, Nika and coworkers analyzed the impact of changing the membrane anchor of Lck with the anchors from other membrane proteins, such as LAT, CD4 and palmytoilation-defective CD4 or CD45
[101][104]. Unexpectedly, only small differences in the ratio of active Lck were observed after changing its membrane anchor, with the exception of the transmembrane segment of CD45, which severely decreased enzymatically active Lck, probably due to the augmented lateral proximity between Lck and CD45, which increased the dephosphorylation of Y394 by endogenous CD45. Collectively, these data suggest that the proximity at the plasma membrane between CD45 and Lck (and thus the activation of the latter) is regulated by the lipids surrounding both proteins. More support for this model comes from molecular dynamics simulations of full-length Lck open and closed conformations using data available from different crystallographic studies, suggesting that Lck interacts with lipids differently in the open and closed Lck conformations, demonstrating that lipid interaction can potentially regulate Lck’s conformation and, in turn, modulate T-cell signaling
[102][105]. This is concordant with the fact that the co-clustering of Lck and the ζ-chain allows for the phosphorylation of ITAMs, even at very high CD45 densities
[42]. Progress in this field involves detailed analyses of the boundary lipid composition of Lck and CD45, although this still remains a difficult technical challenge.
With respect to Lck activity regulation, it has to be considered that it binds to CD4 and CD8 co-receptors through an ion-dependent mechanism involving cysteine residues at positions 20 and 23. In spite of the different immunological functions of cytotoxic CD8+ and helper CD4+ T cells, the TCR-signaling pathways are very similar in both cell populations. Although many approaches have been used to analyze the relevance of co-receptors in Lck activity, it remains unclear whether Lck has a functionally equivalent role in both CD4+ and CD8
[103][106]. Very recently, Horkova et al. generated knockin mice expressing a Lck isoform bearing Cys-to-Ala mutations at positions 20 and 23 (
LckCA) to address the relevance of the interaction between Lck and CD4/CD8 co-receptors
[104][107]. It was previously shown that Lck-co-receptor interaction is needed for the co-receptor–LCK interactions in the positive selection of T cells
[105][108]. In their recent report, Stepanek and coworkers demonstrate that, although Lck
CA molecules are able to transduce pre-TCR and TCR signaling, Lck bound to CD4 and CD8 drives T-cell development and effector immune responses via qualitatively different mechanisms. For example, CD8-bound Lck is not required for antiviral and antitumor activity of cytotoxic T cells in mice, although it enables CD8+ T cell responses to suboptimal antigens. By contrast, the binding of Lck to CD4 is needed for the proper development and function of helper T cells. Very interestingly, differences were observed in the response to high- and low-affinity antigens. The differential role of co-receptor-bound Lck in the response to high- and low-affinity antigens sheds new light on the regulation of TCR signaling and should facilitate the rational design of T-cell-based immunotherapies.
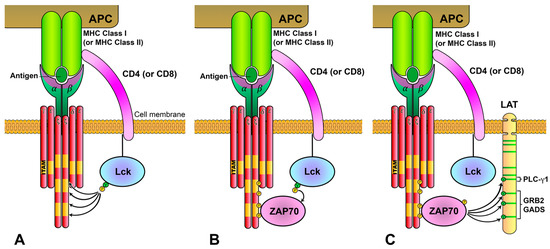
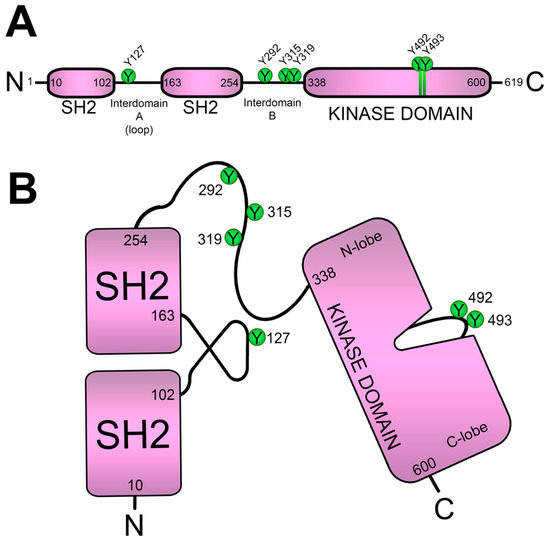
7. Keeping ZAP70 in Check
Lck activity leads to ITAMs phosphorylation in CD3 and the ζ-chain, and doubly phosphorylated ITAMs recruit ZAP70 kinase through its tandem SH2 domains. The binding to phosphorylated ITAMs triggers the opening of the ZAP70 conformation, allowing the phosphorylation of various tyrosine residues. As already mentioned, phosphorylation at Y492 and Y493 stimulates its kinase activity, whereas that at residues Y292, Y315 and Y319 serve to regulate its signaling functions. Indeed, Tyr-to-Phe mutation of residue 319 (
ZAP70Y319F) dramatically impairs LAT and SLP-76 tyrosine phosphorylation, NF-AT activation and IL-2 production
[106][109]. Shortly thereafter, the same group demonstrated that Y319 in the interdomain B of ZAP70 mediates the SH2-dependent interaction between Lck and ZAP70
[107][110]. Tyr to Phe mutation at this residue impeded ZAP70-Lck interaction, giving rise to a molecular explanation for the block in the intracellular signaling of the
ZAP70Y319F mutation. Strikingly, the amino acid sequence encompassing Y319 (YDSP) interacted with the Lck-SH2 domain with a lower affinity than an alternative phosphopeptide containing the sequence YEEI. In their report, Acuto and his colleagues expressed in Jurkat cells a gain-of-function mutant of ZAP70 by changing the sequence Y319SDP into Y319EEI, which caused a prominent increase in TCR-associated intracellular signals, even though its enzymatic activity and binding capacity to Lck were similar to the normal form of ZAP70, unveiling that the Y319-mediated binding to the SH2 domain of Lck is crucial for the propagation of the signaling cascade leading to T-cell activation. Concordantly, in a transgenic mouse model in which the ZAP70 gain-of-function (ZAP-YEEI) mutant was expressed, CD8 T cells showed that ZAP-YEEI expression produced an increase in basal LAT and Erk phosphorylation, as well as at short times (30 s) after stimulation with anti-CD3 antibody
[108][111]. However, at longer times TCR-dependent intracellular signals and IFN-γ production were decreased in CD8 T cells from Tg-ZAP-YEEI mice, suggesting that other downstream negative-feedback mechanisms were being activated in an increased manner. Together with experiments performed with P116 ZAP70-deficient Jurkat cells
[109][112], these data suggest a model in which the Lck-mediated phosphorylation of Y315 and Y319, in addition to the recruitment of downstream activatory and inhibitory effector molecules, also relieves an autoinhibitory intrinsic interaction, allowing for its kinase activity.
Consistent with the important role of tyrosines in the interdomain B of ZAP70, mutation of some of these residues to phenylalanine drastically affected thymic development
[110][111][113,114]. Indeed, Y315F and Y319F mutations attenuated positive and negative selection
[110][111][113,114], while the Y292F mutation upregulated proximal TCR-signaling events. The negative regulatory role played by Y292 residue is probably mediated via its ability to recruit the Cbl and Cbl-b ubiquitin–protein ligases. Along this line, the ZAP-Y292F mutation diminished the internalization and degradation of the TCR/CD3 complex in response to antigenic stimulation, and Cbl recruitment to the immune synapse was also retarded in ZAP-Y292F T cells
[112][115]. Similarly, the mutation of both Y315 and Y319 tyrosines via alanine residues (YYAA) in mice impaired T-cell development and, strikingly, favored the development of rheumatoid factor antibodies, but failed to develop autoimmune arthritis
[113][116]. The crystal structure of ZAP70 confirmed the relevance of tyrosine residues in the interdomain B, showing that Y315 participates in a hydrophobic interaction with W131 in interdomain A, and Y319 interacts with the N-lobe of the catalytic domain
[49][114][115][49,117,118]. Accordingly, Trp-to-Ala mutation (W131A mutation) results in increased responses to TCR stimulation in Jurkat cells
[114][117]. Although
ZAP70W131A knockin mice exhibited relatively normal T-cell development, the crossing to OT II TCR transgenic mice unveiled an increase in the negative selection of OT II+ thymocytes, and also in the numbers of T regulatory cells
[116][119]. Interestingly,
ZAP70W131A mice also showed increased expression of several anergy-related genes.
It is of interest to note that the kinase activity of ZAP70 triggers negative regulatory loops. Taking advantage of a ZAP70 mutant (ZAP70-AS) that can be efficiently and specifically inhibited
[117][120], Arthur Weiss and coworkers used mass spectrometry and phosphoproteomics to analyze the differential pattern of phosphosites in response to ZAP70 inhibition
[118][121]. In their report, they show that, besides the expected reduction in the phosphorylation of downstream substrates, ZAP70 kinase activity inhibition induces increased phosphorylation of ITAMs and Y394 of Lck, and this inhibitory loop seems to be mediated by the phosphorylation of Y192 in Lck (see above).
Other mechanisms have been shown to regulate ZAP70 activity. It has been shown that PKCθ directly interacts with Y127 of ZAP70, and that the interdomain residues Y315 and Y319 are also needed for PKCθ-ZAP70 binding
[119][122]. Interestingly, introducing mutations in PKCθ that block phospho-tyrosine binding prevents not only PKCθ-dependent signaling events, such as nuclear factor κB (NF-κB) activation, but also the phosphorylation of LAT or PLC-γ1, signaling proteins that are traditionally considered to be activated independently of PKC. In addition, it has been proposed that cysteine 564, located in the kinase domain of ZAP70, is palmitoylated, and that a Cys-to-Arg mutation preventing palmitoylation blocks TCR signaling and T-cell activation
[120][123]. However, it has been recently shown that, although such a mutation prevents palmitoylation, a ZAP70 mutant in which cysteine 564 is replaced by a non-palmitoylable alanine residue is able to transduce TCR signaling, and even enhances the activity of Lck and increases its proximity to the TCR
[121][124]. Therefore, C564 seems to be another relevant component in the regulation of proximal TCR signaling by ZAP70 kinase. The generation of animal models with this mutation will certainly be helpful to better understand the biological implications of this regulatory node.
8. Negative-Feedback Loops Involving LAT
Different approaches for the analysis of TCR early events have shown a transitory interaction between the open active form Lck and the LAT adaptor that could downregulate the kinase activity of Lck, and this interaction seems to require a conserved stretch of negatively charged amino acids between residue 113 and residue 126 of LAT
[122][123][125,126]. The substitution of this segment with an uncharged segment has a dual role transmitting TCR incoming signals, increasing proximal signaling events, but leading to a reduction in more downstream signaling events, such as Ca
2+ influx generation or Erk phosphorylation
[124][127]. In 2019, the Arthur Weiss group shed more light on this point, proposing a model in which Lck plays a role as an adaptor molecule indirectly bridging ZAP70 with its substrate LAT
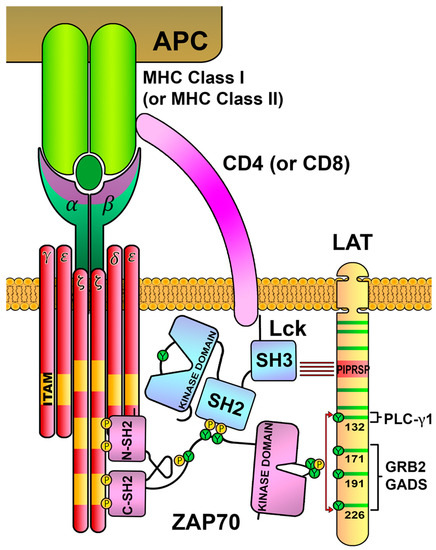
[125][128]. In their report, Lo et al. show that Lck associates with a conserved proline-rich motif (PIPRSP) in LAT via its SH3 domain, and with phospho-ZAP70 via its SH2 domain, thereby acting as a molecular bridge that facilitates the colocalization of ZAP70 and LAT (
Figure 8). Indeed, the elimination of this proline-rich motif (LAT-AIRSA mutant) totally abrogates LAT phosphorylation and other downstream TCR signals. Although a KI mouse model in which these LAT prolines are mutated has not been generated, authors have performed the analysis of thymic development using LAT-deficient hematopoietic stem cells lentivirally transduced to express wild-type LAT or the LAT-AIRSA mutant, showing that this mutation causes a partial block in T-cell development. Therefore, these data demonstrate that Lck is a key player not only in the recruitment and activation of ZAP70, but also in allowing the location of the “active” form of ZAP70 in the proximities of the LAT adaptor through temporally limited interactions, making LAT–Lck interaction a possible mechanism to avoid the prolonged transmission of activatory signals.
Figure 8. Interactions connecting Lck, ZAP70 and LAT. The SH2 domain of Lck binds to the phosphorylated tyrosines in the interdomain B of ZAP70, and the SH3 domain of Lck binds to the proline-rich region of LAT (PIPRSP). This binding bridge generated by Lck localizes the ZAP70 kinase close to the LAT adaptor, favoring its tyrosine phosphorylation.
In the course of immune responses, antigen-specific T cells are activated and first undergo a phase of clonal expansion, followed by a contraction phase that restores normal T-cell numbers and requires apoptosis. Although Fas engagement is essential for T-lymphocyte apoptosis and immune-system homoeostasis, it has been demonstrated that the simultaneous engagement of TCR, CD28 and Fas in naive human T lymphocytes led to decreased activation and proliferation
[126][129]. This proteolytic cleavage generates N-terminal truncated forms of LAT that lack the essential C-terminal tyrosines but still retain the proline-rich region and the negatively charged segment, and theoretically would still be capable of Lck binding. The proteolytic cleavage of other adaptors with intracellular-signaling functions has also been demonstrated
[127][128][129][131,132,133]; thus, this may be a general mechanism of the termination of intracellular signals coupled to immune receptors. In this regard, a family has been described in which mutations in LAT resulting in the generation of a premature stop-codon and protein truncation leads to a novel form of severe combined immunodeficiency
[130][134]. This mutation generated a form of LAT truncation shortly after the transmembrane domain, and the affected individuals had extremely low numbers of CD4 and CD8 T cells, consistent with the impaired T-cell development observed in LAT-deficient murine models
[71]. Another study in humans describes a homozygous mutation in exon 5, leading to a premature stop codon deleting most of the cytoplasmic tail of LAT, and presenting a more complicated phenotype with some unexpected clinical features
[131][135]. In their work, Keller et al. show that three patients homozygous for this mutation, which generates a truncated form of LAT lacking the critical C-terminal tyrosine residues for signal propagation, presented with combined immunodeficiency and severe autoimmune disease from early childhood. Contrasting the
Lat4YF knockin mice (in which the last four C-terminal tyrosines were mutated to phenylalanine residues)
[73], reduced numbers of T cells were present in the patients, and they were able to induce Ca
2+ influx and nuclear factor (NF) kappa B signaling, unveiling unpredicted functions for the N-terminal region of LAT preceding the four C-terminal tyrosine residues, which could play a regulatory role to be analyzed in more detail.
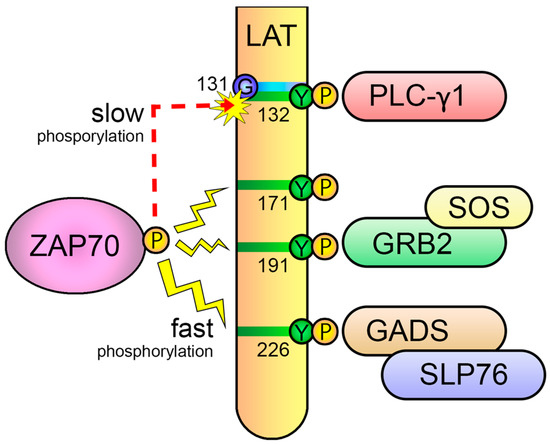
Another point of the negative regulation of TCR signaling appears to be localized to the sixth tyrosine of LAT (human number 132, mouse number 136). This residue has the exclusive ability to recruit PLC-γ1, which is crucial for Ca
2+ mobilization, Erk and PKC activation, and eventually the irreversible activation of T cells
[132][136]. However, phosphorylation kinetics of this residue is slow compared to other phosphorylation sites on LAT
[133][137]. The slow phosphorylation kinetics of this relevant tyrosine residue is due to the presence of an evolutionarily conserved glycine just preceding the tyrosine, which makes it a poor substrate for ZAP70, as the kinase domain of ZAP70 strongly prefers an acidic residue (aspartate or glutamate) at the −1 position relative to the substrate tyrosine residues
[134][138]. This slow kinetics suggested a molecular mechanism to support the kinetic-proofreading (KPR) model of TCR ligand discrimination
[135][139] (
Figure 9). This model proposes that, after TCR-pMHC binding, there is a time lag between TCR engagement and irreversible T-cell activation, and, consequently, the longer the pMHC-TCR interaction, the better the T-lymphocyte activation. In 2019, Lo et al. examined the effects of replacing the glycine residue at position 131 (G131) in the human form of LAT with aspartate (LAT-G131D) or glutamate (LAT-G131E), and found that this leads to a remarkable and specific increase in LAT-Y132 phosphorylation, PLC-γ1 phosphorylation and Ca
2+ influx generation
[136][140]. Moreover, they found that the difference between wild-type LAT and the LAT-G131D or LAT-G131E mutant increase is remarkably higher when cells are stimulated with low-affinity peptides, supportive of the proposal of an important role for the phosphorylation kinetics of the sixth tyrosine residue of LAT in the discrimination between self and non-self. A recent report, in which mass spectrometry-based methods were used to quantify the first molecular events triggered after TCR stimulation with peptides of varying affinities has lent support to this model
[137][141]. In their work, Roncagalli and coworkers show that differences in the signaling between strong ligands and weak ligands was not reflected by changes in very early signaling events, but rather by changes in later events focused around ZAP70 activation and the phosphorylation of the signaling adaptor LAT.
Figure 9. Slow phosphorylation of the sixth tyrosine of LAT regulates TCR signaling. The evolutionarily conserved glycine residue preceding the sixth tyrosine of LAT (Y132 in human LAT) makes it a worse substrate for ZAP70, leading to a slower phosphorylation of this residue with regard to the other C-terminal tyrosines. This slow kinetics phosphorylation is a molecular mechanism that supports the kinetic-proofreading (KPR) model of TCR ligand discrimination.
AOur group also contributed to give support to this model, as well as to unveil the potential of accelerating Ca
2+ influx generation to regulate peripheral tolerance
[138][142]. Besides verifying that a LAT
G131D mutant increased Y132 phosphorylation and Ca
2+ fluxes in Jurkat cells,
rwe
searchers showed that cells expressing the LAT
G131D mutant secrete greater amounts of interleukin-2 (IL-2) in response to CD3/CD28 engagement, but cells expressing the LAT
G131D mutant were more sensitive to the inhibition of IL-2 production via pre-treatment with anti-CD3, which points to a possible role of this residue in the generation of anergy.
RWe
searchers and others have generated a knockin strain of mice in which the glycine residue preceding the sixth tyrosine residue (Y136 in mice) was substituted by an aspartate (
LatG135D mice)
[139][140][143,144]. Consistent with the in vitro results, T cells from
LatG135D mice showed increased peripheral T-cell activation and proliferation, a larger number of anergic T cells, fewer peripheral CD8 T cells and more γδ-T cells than their wild-type littermates. Remarkably, the LAT
G135D mutation is dominant, as heterozygous mice also showed an altered phenotype compared to wild-type mice
[139][143]. Also of interest is the fact that
LatG135D mice showed enhanced self-reactivity, which increased thymic negative selection
[140][144]. Moreover, Lo et al. performed
Listeria infection experiments, showing that T cells from
LatG135D mice proliferate more than their wild-type counterparts in response to very weak stimuli, but display an imbalance between effector and memory responses. Strikingly, furthermore, despite increased negative selection and anergy, mice expressing the LAT
G135D mutant exhibit features associated with autoimmunity and immunopathology. Altogether, these results show that the phosphorylation of LAT at Y136 constitutes an important step for the kinetic-proofreading model, which requires the proper conditions, as both signaling deficiency and hyperactivity can lead to immunodeficiency and/or autoimmunity.