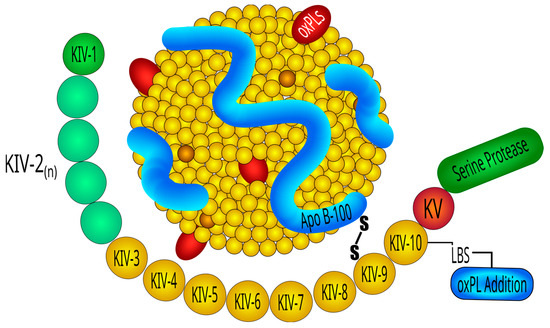
2.3. Lp(a) Metabolism
Lp(a) biosynthesis faces four main steps: transcription of LPA, protein translation, transfer to the secretory pathway, and assembly of the Lp(a) particles. Lp(a) is exclusively produced in the liver which secretes apo(a)- and apoB-containing lipoproteins separately, so that the final assembly of Lp(a) takes place extracellularly by covalent linkage of apo(a) with apoB
[87][123]. Synthesis and secretion are regulated by the effects of genetic control of LPA expression and processing of the apo(a) protein, respectively.
Catabolism of Lp(a) is not entirely clear. Regulation and function of the endocytic receptor which removes Lp(a) from the circulation is still a matter of debate
[88][125]. The presence of apo(a) and perhaps also involvement of proprotein convertase subtilisin/kexin type 9 (PCSK9) limits removal of Lp(a) by the LDL-receptor. In addition, several other endocytic receptors have been implicated to mediate removal of Lp(a) from blood, including LDL-receptor related protein 1, very low density lipoprotein receptor, scavenger receptor B1, and plasminogen receptor KT (PlgRKT)
[88][89][125,126]. While most lipoprotein receptors direct Lp(a) into a route which leads to the lysosomal degradation of the entire particle, PlgRKT was reported to shuttle Lp(a) into a pathway which leads to the selective degradation of the lipids and apoB, but to the re-secretion of apo(a) which then associates with another LDL-particle to form a new Lp(a) particle.
2.4. Distribution of Lp(a) Concentration and Effects of Non-Genetic Factors
The distribution of plasma Lp(a) levels is highly variable among different ethnic groups with concentrations varying up to 1000-fold within each population, ranging from less than 0.1 mg/dL to as high as 387 mg/dL. The lowest levels are seen in non-Hispanic Caucasians, Chinese, and Japanese; slightly higher levels have been documented in Hispanics, while the highest levels are found in Blacks [90][127]. In Caucasians, plasma levels are comparable in men and women, and it is estimated that 20% of the population worldwide has an Lp(a) level >50 mg/dL (>105 nmol/L) [91][128], 5% of individuals has an Lp(a) level above 120 mg/dL (250 nmol/L), whereas only 1% of individuals has an extremely elevated Lp(a) level above the 99th percentile, corresponding approximately to 180 mg/dL. Plasma levels are generally unaffected by dietary interventions or various physiological and environmental factors, including age, sex, fasting state, or physical activity, but are also known to be slightly influenced by pregnancy, menopause, hormone use, cholestasis, thyroid dysfunction, acute phase events, and renal function [92][129].
2.5. Lp(a) Measurement
Reproducible and reliable measurement of Lp(a) was difficult to obtain mainly because of the highly polymorphic nature of the apo(a) moiety, due to the variation in isoform size. Additional factors included lack of standardization across laboratories with some assays reporting Lp(a) values as mass concentrations (mg/dL) and others as particle concentrations (nmol/L)
[93][131], and the adoption of antibody-based approaches which led to possible underestimation of the small isoforms and overestimation of the large isoforms
[55]. The efforts of The International Federation of Clinical Chemistry and Laboratory Medicine to standardize reference material to calibrate Lp(a) assays improved the reproducibility between methods
[94][132], although some degree of heterogeneity might persist
[95][133].
3. Plasma Lp(a) Concentrations in Hypertension
Dyslipidemia is more prevalent in hypertensive than normotensive individuals, and changes in lipid levels progressively worsen with increasing BP
[96][135]. Increased levels of total and low-density lipoprotein cholesterol and triglycerides, and lower high-density lipoproteins cholesterol were reported in hypertensive patients
[97][136]. Regarding Lp(a), data are highly controversial mostly depending upon lack of standardization of assays and relevant differences among ethnic groups. While some studies reported higher Lp(a) concentrations in hypertensive than normotensive subjects, other studies did not
[98][99][100][101][102][103][104][105][106][107][108][65,137,138,139,140,141,142,143,144,145,146]. In a study conducted by Lip et al. on ambulatory hypertensive patients, median Lp(a) levels were found to be markedly elevated in Blacks, in line with previous observations
[106][144]. Elevated plasma Lp(a) was also more frequent in hypertensive patients of Indian than Caucasian descent, although in hypertensive Caucasians no differences were observed with the respective normotensive subjects. In agreement with these findings, two additional studies reported an increased prevalence of elevated Lp(a) in Indian hypertensives free of cardiovascular complications in comparison to their respective normotensive controls
[109][110][147,148].
4. Lp(a) and The Vascular Wall
Essential hypertension is the most frequent form of hypertension and is characterized by a complex and multifactorial pathophysiology, where blood vessels, heart, and kidneys are reciprocally involved in regulation of the leading determinants of systemic BP, namely cardiac output and peripheral vascular resistance
[111][149]. Within this complex interplay, a crucial role belongs to vascular endothelium that, in normal conditions, balances vasoconstriction and vasodilation of resistance vessels, also exerting important antithrombotic and anti-inflammatory functions that might be impaired in the process of atherogenesis
[112][150].
In vitro studies indicate that elevated Lp(a) can directly contribute to atherogenesis and cause endothelial cell (ECs) and vascular smooth muscle cell (VSMCs) dysfunction. These effects appear to be prevalently mediated by the apo(a) moiety, due to its hydrophilic properties which allow a direct interaction with the vascular endothelium as well as other cellular receptors
[113][154]. Much like other lipoproteins, Lp(a) can diffuse passively through endothelial surfaces via concentration gradients, accumulating in subendothelial spaces where, after binding to proteoglycans and other subendothelial structures, becomes oxidized, promotes inflammation, and mediates atherogenesis
[114][155]. Lp(a) accumulation and retention in the vessel wall and sub-endothelial surfaces is facilitated by a potent lysine-binding pocket present on K4 type-10 that binds to exposed lysine on denuded endothelial surfaces and to components of the subendothelial matrix
[115][156].
In animal models, high Lp(a) levels impair endothelium-dependent vasodilation, as demonstrated by a dose-dependent reduction in the expression of inducible nitric oxide synthase, both at mRNA and protein level
[116][117][167,168]. Lp(a) can also affect vascular smooth muscle cells (VSMCs), as suggested by early in vitro studies showing that cell migration and proliferation could be triggered by apo(a)-mediated downregulation of plasmin-dependent activation of transforming growth factor-β
[118][169].
5. Lp(a) and Organ Damage in Hypertension
The development of organ damage in essential hypertensive patients is likely related, but not limited to, the direct effects of increased BP levels. Additional factors, including circulating lipids, play a crucial role in the process of vascular damage since dyslipidemia, which is frequently detected in hypertension, and serum levels of specific lipoproteins greatly affect cardiovascular morbidity and mortality in hypertensive populations
[119][179]. Retrospective and prospective studies have shown that high serum levels of Lp(a) are an independent risk factor for cardiovascular diseases
[120][180].
Arterial vessels are one of the main targets of hypertension and a broad interest has gathered around the mechanisms that might contribute to arterial stiffening. Arterial stiffness is recognized as a strong predictor of cardiovascular events, both in the general population
[121][122][185,186] and in hypertensive patients
[123][124][187,188]. Currently, arterial stiffness can be estimated by noninvasive methods, including measurements of the augmentation index (AIx) by pulse wave analysis and carotid–femoral pulse wave velocity (PWV), which provide a comprehensive assessment of the stiffness of the entire arterial tree (elastic plus muscular arteries and arterioles)
[125][189]. These are now widely accepted tools for the assessment of subclinical arterial damage in hypertension
[126][61]. Increased arterial stiffness has been associated with major cardiovascular risk factors, including being overweight or obese, impaired glucose tolerance, dyslipidemia, and smoking
[127][128][129][130][190,191,192,193] and is characterized by both structural and functional changes of the vascular wall
[131][194].
The hypothesis of a possible contribution of Lp(a) to arterial stiffening was initially investigated in elderly Japanese subjects with type 2 diabetes, reporting an independent association of its levels with the PWV
[132][198]. A significant correlation between the plasma levels of oxidized Lp(a) and PWV was also reported in a subset of relatively old (mean age: 66 years) hypertensive patients with coronary artery disease and diabetes
[133][199]. Similarly, in a small study enrolling hypertensive women, a significant relationship was reported between oxidized Lp(a) levels and the cardio–ankle vascular index
[134][200].
All these findings might have considerable clinical implications for the identification of target organ damage in hypertensive patients as well as for their prevention and treatment. Lp(a) measurement might indeed be useful in the diagnostic workup of these patients, to identify those who might be more prone to developing organ damage. On the other hand, in consideration of the encouraging data coming from clinical trials of new Lp(a)-lowering therapies, in the near future, a reduction in Lp(a) levels might possibly improve hypertension outcomes.
6. Lp(a) and Hypertensive Renal Damage
Despite robust evidence suggesting an inverse relationship between renal function and plasma Lp(a) levels in patients with severely impaired glomerular filtration rate
[16][135][16,201], only a few studies have investigated this relationship in patients with hypertensive nephrosclerosis. Data obtained from large cohorts of subjects with end-stage renal disease that included mostly patients with diabetic nephropathy suggested a reciprocal relationship between Lp(a) levels and renal function.
In a cross-sectional study of 417 hypertensive patients, 160 of whom had glomerular filtration rate from 30 to 89 mL/min/1.73 m
2 of body surface area, scholars measured serum lipids and apolipoproteins
[136][130]. Lp(a) levels were significantly higher in patients with early impairment of renal function caused by hypertensive nephrosclerosis and, most importantly, there was a highly significant inverse relationship between Lp(a) levels and glomerular filtration rate.
7. Lipoprotein(a): Dietary and Pharmacological Interventions
Regarding lifestyle interventions, there is a historical assumption according to which diet has no effect on Lp(a). However, since the first report of dietary effects on Lp(a) in 1991
[137][210], there have been only a few well controlled clinical studies investigating the consequences of dietary modification on its levels. Overall, current evidence, albeit limited, indicates that diet modulates only modestly Lp(a) and often in the opposite direction to LDL-C
[138][139][140][141][142][143][144][145][146][211,212,213,214,215,216,217,218,219].
The resurgence of Lp(a) in the context of cardiovascular prevention and treatment is mainly due to the relatively recent development of new RNA-directed treatments. Indeed, traditional lipid-lowering agents had demonstrated little and clinically irrelevant effects on Lp(a). These agents included statins; niacin (20% reduction of Lp(a) at maximal dose but with an unknown effectiveness in reducing major atherosclerotic cardiovascular events-MACE)
[147][221]; cholesteryl ester transfer protein (20–30% reduction of Lp(a) with no reduction in MACE)
[148][222]; and apolipoprotein B100 antisense oligonucleotide mipomersen (20–40% reduction in Lp(a) levels but MACE reduction unknown in patients with elevated Lp(a) with significant concerns regarding potential hepatotoxicity which justifies limited approval for homozygous familiar hypercholesterolemia)
[149][223].
8. Conclusions
The relationship between elevated Lp(a) levels and essential hypertension is still a matter of intriguing debate due to limited clinical evidence in support of a causal and/or reciprocal association. Nevertheless, solid evidence indicates that elevated Lp(a) levels can significantly contribute to cardiovascular and renal damage in hypertensive patients, leading to a worse clinical outcome. These effects of Lp(a) could be ascribed to the multiple detrimental effects that Lp(a) exerts on the vascular wall. Recent evidence of a longitudinal relationship of Lp(a) levels with the cardiovascular outcome in a large multiethnic cohort of hypertensive patients reinforces the need for more extensive clinical research in this field. This is further encouraged by the very promising results of the studies that have employed novel RNA-targeted treatments for Lp(a) reduction. If their benefits will be confirmed by the ongoing outcome trials, these new treatments will shift the gear for effective intervention on the residual cardiovascular risk of patients with high BP.