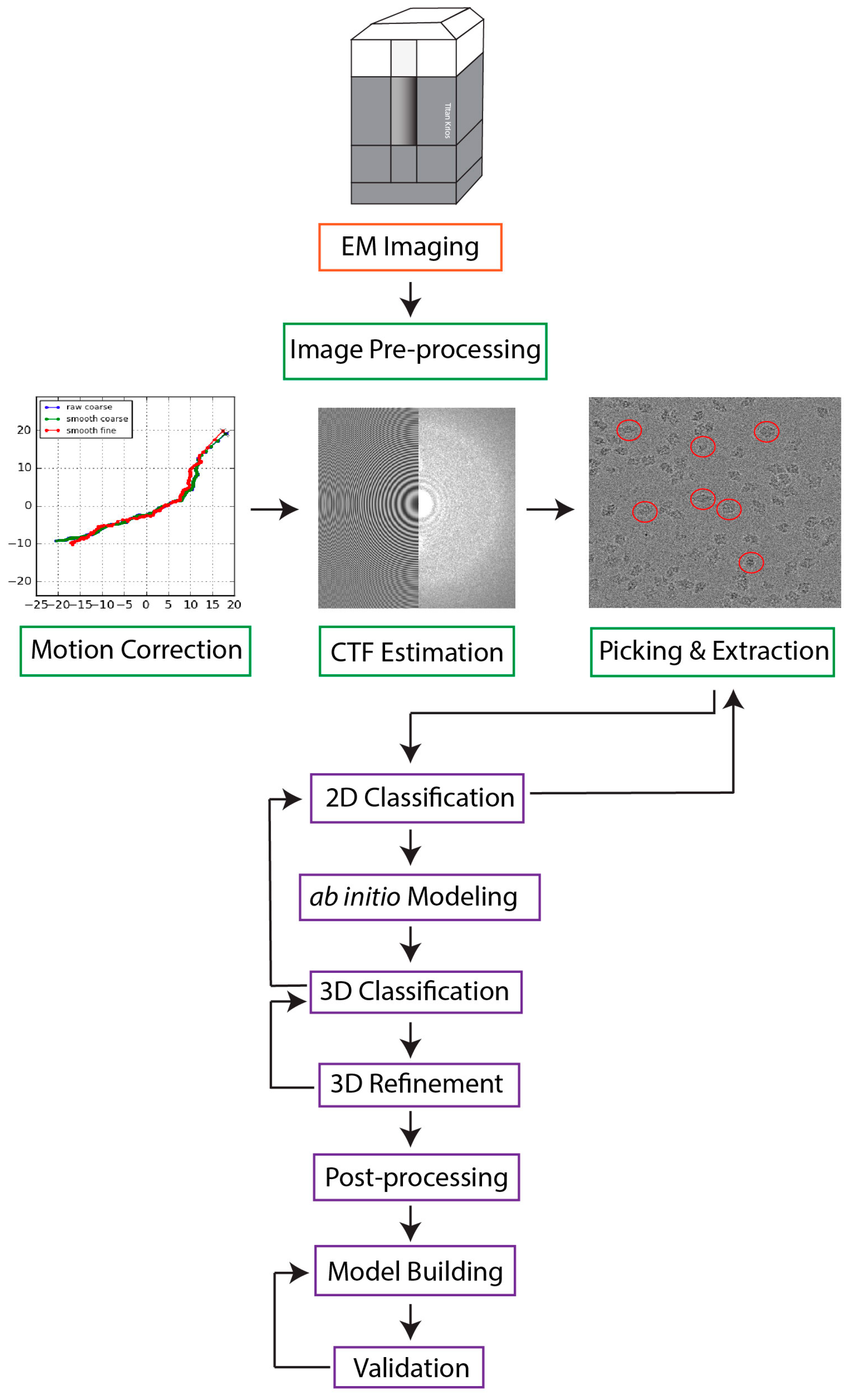
Single particle cryo-electron microscopy (cryo-EM) has emerged as the prevailing method for near-atomic structure determination, shedding light on the important molecular mechanisms of biological macromolecules. However, the inherent dynamics and structural variability of biological complexes coupled with the large number of experimental images generated by a cryo-EM experiment make data processing nontrivial. In particular, ab initio reconstruction and atomic model building remain major bottlenecks that demand substantial computational resources and manual intervention. Approaches utilizing recent innovations in artificial intelligence (AI) technology, particularly deep learning, have the potential to overcome the limitations that cannot be adequately addressed by traditional image processing approaches.
- cryo-electron microscopy
- cryo-EM
- deep learning
- machine learning
- artificial intelligence
- AI
- neural networks
1. Introduction
2. Artificial Intelligence-Based Approaches to De Novo Model Building
| Program | AI Architecture | Advantages | Limitations |
|---|---|---|---|
| Emap2Sec [34] | CNN | Identifies SSEs Demonstrated on experimental maps High accuracy for intermediate resolution maps |
Does not place α-helices and β-sheets in detected regions Voxel-based approach fails for large EM maps |
| Emap2Sec+ [36] | ResNet | Identifies SSEs and nucleic acids in maps 5–10 Å Improved accuracy in protein SSE detection compared to Emap2Sec [34] |
Voxel-based approach fails for large EM maps |
| CNN-based [35] | CNN | Identifies SSEs | Does not place α-helices and β-sheets in detected regions No results for experimental maps |
| Haruspex [49] | U-Net | Identifies SSEs and nucleic acids in maps Applied to experimental and simulated maps |
Only applicable to maps ≤4 Å False positives for helices, sheets or RNA/DNA Misclassifies semi-helical elements, β-hairpin turns, polyproline residues as α-helices |
| EMNUSS [50] | U-Net | Identifies SSEs Applied to experimental and simulated maps |
Incorrect predictions on atypical density volumes; narrow receptive field |
| AAnchor [40] | CNN | Identifies amino acids | Only applicable to maps ≤3.1 Å Only detects 10–20% amino acids on average |
| A2-Net [41] | 3D-CNN, MCTS | Identifies amino acids Model with 1000 amino acids can be derived in minutes Applied to experimental maps Fully automated |
Cannot identify ligands |
| Structure Generator [39] | 3D-CNN, GCN, LSTM |
Identifies amino acids and rotamer orientation, builds full protein chain | Limited to protein sequences <700 amino acids No results for experimental maps |
| CR-I-TASSER [38] | 3D-CNN, I-TASSER | 3D-CNN predicts Cα atoms used for I-TASSER; generates full structure | Cα trace prediction accuracy dependent on resolution Requires prior map segmentation |
| DeepTracer [51][59] | 3D U- Net |
Locates amino acid positions and protein backbone Identifies SSEs and amino acids Automated Lastest version models nucleic acids |
Only applicable to maps ≤5 Å Only builds atoms for main chains |
| DeepTracer ID [64] | 3D U-Net, AlphaFold2 | Uses DeepTracer to build model and searches against AlphaFold2 library for refinement Identifies individual proteins in density map; does not require protein sequences to be known a priori Does not require high accuracy from AlphaFold2 prediction |
Limited to proteins >100 amino acids for succesful AlphaFold2 prediction matches Only applicable to maps ≤4.2 Å |
| EMBUILD [52] | U-Net, AlphaFold2 | Constructs main chain map; fits AlphaFold2 predicted chains into the map | Only builds atoms for main chains |
| ModelAngelo [61][62] | CNN, GNN | Builds complete atomic model Better RMSD and sequence prediction results than DeepTracer |
Only applicable to maps ≤3.5 Å Requires protein sequences are known |
| DeepMM [65] | CNN | Predicts Cα positons; identifies SSEs and amino acids Applied to experimental maps |
Cannot model nucleic acids Residue matching accuracy dependent on map resolution |
| DEMO-EM [66] | ResNET, I-TASSER | Builds complete atomic model for multi-domain proteins Only requires protein sequence Fully automated |
Requires prior map segmentation Individual domain models calculated without restraints from density data |
3. Applications of Artificial Intelligence-Based Protein Structure Prediction to Atomic Model Building
References
- DiIorio, M.C.; Kulczyk, A.W. Exploring the Structural Variability of Dynamic Biological Complexes by Single-Particle Cryo-Electron Microscopy. Micromachines 2022, 14, 118.
- DiIorio, M.C.; Kulczyk, A.W. A Robust Single-Particle Cryo-Electron Microscopy (cryo-EM) Processing Workflow with cryoSPARC, RELION, and Scipion. J. Vis. Exp. 2022, 179, e63387.
- Kulczyk, A.W.; Moeller, A.; Meyer, P.; Sliz, P.; Richardson, C.C. Cryo-EM structure of the replisome reveals multiple interactions coordinating DNA synthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E1848–E1856.
- Kulczyk, A.W.; Sorzano, C.O.S.; Grela, P.; Tchorzewski, M.; Tumer, N.E.; Li, X.P. Cryo-EM structure of Shiga toxin 2 in complex with the native ribosomal P-stalk reveals residues involved in the binding interaction. J. Biol. Chem. 2023, 299, 102795.
- Burley, S.K.; Berman, H.M.; Chiu, W.; Dai, W.; Flatt, J.W.; Hudson, B.P.; Kaelber, J.T.; Khare, S.D.; Kulczyk, A.W.; Lawson, C.L.; et al. Electron microscopy holdings of the Protein Data Bank: The impact of the resolution revolution, new validation tools, and implications for the future. Biophys. Rev. 2022, 14, 1281–1301.
- Kulczyk, A.W.; McKee, K.K.; Zhang, X.M.; Bizukojc, I.; Yu, Y.Q.; Yurchenco, P.D. Cryo-EM reveals the molecular basis of laminin polymerization and LN-lamininopathies. Nat. Commun. 2023, 14, 317.
- Kuhlbrandt, W. The resolution revolution. Science 2014, 343, 1443–1444.
- Earl, L.A.; Falconieri, V.; Milne, J.L.; Subramaniam, S. Cryo-EM: Beyond the microscope. Curr. Opin. Struct. Biol. 2017, 46, 71–78.
- Bai, X.C.; McMullan, G.; Scheres, S.H.W. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57.
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245.
- Fica, S.M.; Nagai, K. Cryo-electron microscopy snapshots of the spliceosome: Structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol. 2017, 24, 791–799.
- Wong, W.; Bai, X.C.; Brown, A.; Fernandez, I.S.; Hanssen, E.; Condron, M.; Tan, Y.H.; Baum, J.; Scheres, S.H. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife 2014, 3, e03080.
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296.
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530.
- de la Rosa-Trevin, J.M.; Quintana, A.; Del Cano, L.; Zaldivar, A.; Foche, I.; Gutierrez, J.; Gomez-Blanco, J.; Burguet-Castell, J.; Cuenca-Alba, J.; Abrishami, V.; et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 2016, 195, 93–99.
- Grant, T.; Rohou, A.; Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 2018, 7, e35383.
- Shaikh, T.R.; Gao, H.; Baxter, W.T.; Asturias, F.J.; Boisset, N.; Leith, A.; Frank, J. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 2008, 3, 1941–1974.
- van Heel, M.; Harauz, G.; Orlova, E.V.; Schmidt, R.; Schatz, M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996, 116, 17–24.
- Ludtke, S.J.; Baldwin, P.R.; Chiu, W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999, 128, 82–97.
- Lander, G.C.; Stagg, S.M.; Voss, N.R.; Cheng, A.; Fellmann, D.; Pulokas, J.; Yoshioka, C.; Irving, C.; Mulder, A.; Lau, P.W.; et al. Appion: An integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 2009, 166, 95–102.
- Baldwin, P.R.; Tan, Y.Z.; Eng, E.T.; Rice, W.J.; Noble, A.J.; Negro, C.J.; Cianfrocco, M.A.; Potter, C.S.; Carragher, B. Big data in cryoEM: Automated collection, processing and accessibility of EM data. Curr. Opin. Microbiol. 2018, 43, 1–8.
- Namba, K.; Makino, F. Recent progress and future perspective of electron cryomicroscopy for structural life sciences. Microscopy 2022, 71, i3–i14.
- Penczek, P.A.; Fang, J.; Li, X.; Cheng, Y.; Loerke, J.; Spahn, C.M. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy 2014, 140, 9–19.
- Heimowitz, A.; Anden, J.; Singer, A. Reducing bias and variance for CTF estimation in single particle cryo-EM. Ultramicroscopy 2020, 212, 112950.
- Arbelaez, P.; Han, B.G.; Typke, D.; Lim, J.; Glaeser, R.M.; Malik, J. Experimental evaluation of support vector machine-based and correlation-based approaches to automatic particle selection. J. Struct. Biol. 2011, 175, 319–328.
- Heimowitz, A.; Anden, J.; Singer, A. APPLE picker: Automatic particle picking, a low-effort cryo-EM framework. J. Struct. Biol. 2018, 204, 215–227.
- Al-Azzawi, A.; Ouadou, A.; Tanner, J.J.; Cheng, J. AutoCryoPicker: An unsupervised learning approach for fully automated single particle picking in Cryo-EM images. BMC Bioinform. 2019, 20, 326.
- Yang, Z.; Fang, J.; Chittuluru, J.; Asturias, F.J.; Penczek, P.A. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 2012, 20, 237–247.
- Ma, L.; Reisert, M.; Burkhardt, H. RENNSH: A novel alpha-helix identification approach for intermediate resolution electron density maps. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 228–239.
- Chen, M.; Baldwin, P.R.; Ludtke, S.J.; Baker, M.L. De Novo modeling in cryo-EM density maps with Pathwalking. J. Struct. Biol. 2016, 196, 289–298.
- Si, D.; Ji, S.; Nasr, K.A.; He, J. A machine learning approach for the identification of protein secondary structure elements from electron cryo-microscopy density maps. Biopolymers 2012, 97, 698–708.
- Giri, N.; Roy, R.S.; Cheng, J. Deep learning for reconstructing protein structures from cryo-EM density maps: Recent advances and future directions. Curr. Opin. Struct. Biol. 2023, 79, 102536.
- Si, D.; Nakamura, A.; Tang, R.B.; Guan, H.W.; Hou, J.; Firozi, A.; Cao, R.Z.; Hippe, K.; Zhao, M.L. Artificial intelligence advances for de novo molecular structure modeling in cryo-electron microscopy. WIREs Comput. Mol. Sci. 2022, 12, e1542.
- Subramaniya, S.R.M.V.; Terashi, G.; Kihara, D. Protein Secondary Structure Detection in Intermediate-Resolution Cryo-EM Maps using Deep Learning. Nat. Methods 2020, 118, 43a.
- Li, R.J.; Si, D.; Zeng, T.; Ji, S.W.; He, J. Deep Convolutional Neural Networks for Detecting Secondary Structures in Protein Density Maps from Cryo-Electron Microscopy. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 41–46.
- Wang, X.; Alnabati, E.; Aderinwale, T.W.; Subramaniya, S.R.M.V.; Terashi, G.; Kihara, D. Emap2sec+: Detecting Protein and DNA/RNA Structures in Cryo-EM Maps of Intermediate Resolution Using Deep Learning. Acta Crystallogr. Sect. A 2021, 77, A84.
- Si, D.; Moritz, S.A.; Pfab, J.; Hou, J.; Cao, R.Z.; Wang, L.G.; Wu, T.Q.; Cheng, J.L. Deep Learning to Predict Protein Backbone Structure from High-Resolution Cryo-EM Density Maps. Sci. Rep. 2020, 10, 4282.
- Zhang, X.; Zhang, B.; Freddolino, P.L.; Zhang, Y. CR-I-TASSER: Assemble protein structures from cryo-EM density maps using deep convolutional neural networks. Nat. Methods 2022, 19, 195–204.
- Li, P.N.; de Oliveira, S.H.P.; Wakatsuki, S.; van den Bedem, H. Sequence-guided protein structure determination using graph convolutional and recurrent networks. In Proceedings of the IEEE 20th International Conference on Bioinformatics and Bioengineering (BIBE), Cincinnati, OH, USA, 26–28 October 2020; pp. 122–127.
- Rozanov, M.; Wolfson, H.J. AAnchor: CNN guided detection of anchor amino acids in high resolution cryo-EM density maps. In Proceedings of the IEEE 20th International Conference on Bioinformatics and Bioengineering (BIBE), Madrid, Spain, 3–6 December 2018; pp. 88–91.
- Xu, K.; Wang, Z.; Shi, J.P.; Li, H.S.; Zhang, Q.C. A(2)-Net: Molecular Structure Estimation from Cryo-EM Density Volumes. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; pp. 1230–1237.
- He, K.M.; Zhang, X.Y.; Ren, S.Q.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 26 June–1 July 2016; pp. 770–778.
- Wang, R.Y.R.; Kudryashev, M.; Li, X.M.; Egelman, E.H.; Basler, M.; Cheng, Y.F.; Baker, D.; DiMaio, F. De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nat. Methods 2015, 12, 335–384.
- Bai, R.; Wan, R.; Wang, L.; Xu, K.; Zhang, Q.; Lei, J.; Shi, Y. Structure of the activated human minor spliceosome. Science 2021, 371, eabg0879.
- Dong, S.; Huang, G.; Wang, C.; Wang, J.; Sui, S.F.; Qin, X. Structure of the Acidobacteria homodimeric reaction center bound with cytochrome c. Nat. Commun. 2022, 13, 7745.
- Liu, W.; Rabinovich, A.; Berg, A.C. ParseNet: Looking wider to see better. arXiv 2015, arXiv:1506.04579.
- Peng, C.; Zhang, X.Y.; Yu, G.; Luo, G.M.; Sun, J. Large Kernel Matters—Improve Semantic Segmentation by Global Convolutional Network. In Proceedings of the 30th IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017; pp. 1743–1751.
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Munich, Germany, 5–9 October 2015; pp. 234–241.
- Mostosi, P.; Schindelin, H.; Kollmannsberger, P.; Thorn, A. Haruspex: A Neural Network for the Automatic Identification of Oligonucleotides and Protein Secondary Structure in Cryo-Electron Microscopy Maps. Angew. Chem. Int. Ed. 2020, 59, 14788–14795.
- He, J.H.; Huang, S.Y. EMNUSS: A deep learning framework for secondary structure annotation in cryo-EM maps. Brief. Bioinform. 2021, 22, bbab156.
- Pfab, J.; Phan, N.M.; Si, D. DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl. Acad. Sci. USA 2021, 118, e2017525118.
- He, J.; Lin, P.; Chen, J.; Cao, H.; Huang, S.Y. Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly. Nat. Commun. 2022, 13, 4066.
- Singh, S.; Vanden Broeck, A.; Miller, L.; Chaker-Margot, M.; Klinge, S. Nucleolar maturation of the human small subunit processome. Science 2021, 373, eabj5338.
- Tan, Y.B.; Chmielewski, D.; Law, M.C.Y.; Zhang, K.; He, Y.; Chen, M.Y.; Jin, J.; Luo, D.H. Molecular architecture of the Chikungunya virus replication complex. Sci. Adv. 2022, 8, eadd2536.
- Porta, J.C.; Han, B.; Gulsevin, A.; Chung, J.M.; Peskova, Y.; Connolly, S.; Mchaourab, H.S.; Meiler, J.; Karakas, E.; Kenworthy, A.K.; et al. Molecular architecture of the human caveolin-1 complex. Sci. Adv. 2022, 8, eabn7232.
- Terashi, G.; Kihara, D. De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun. 2018, 9, 1618.
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221.
- Frenz, B.; Walls, A.C.; Egelman, E.H.; Veesler, D.; DiMaio, F. RosettaES: A sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 2017, 14, 797–800.
- Nakamura, A.; Meng, H.; Zhao, M.; Wang, F.; Hou, J.; Cao, R.; Si, D. Fast and automated protein-DNA/RNA macromolecular complex modeling from cryo-EM maps. Brief. Bioinform. 2023, 24, bbac632.
- Yang, T.J.; Chang, Y.C.; Ko, T.P.; Draczkowski, P.; Chien, Y.C.; Chang, Y.C.; Wu, K.P.; Khoo, K.H.; Chang, H.W.; Hsu, S.D. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc. Natl. Acad. Sci. USA 2020, 117, 1438–1446.
- Jamali, K.; Kimanius, D.; Scheres, S.H. A Graph Neural Network Approach to Automated Model Building in Cryo-EM Maps. In Proceedings of the International Conference on Learning Representations, Kigali, Rowanda, 1–5 May 2023.
- Jamali, K.; Kall, L.; Zhang, R.; Brown, A.; Kimanius, D.; Scheres, S.H. Automated model building and protein identification in cryo-EM maps. bioRxiv 2023.
- Giri, N.; Cheng, J.L. Improving Protein-Ligand Interaction Modeling with cryo-EM Data, Templates, and Deep Learning in 2021 Ligand Model Challenge. Biomolecules 2023, 13, 132.
- Chang, L.C.; Wang, F.B.; Connolly, K.; Meng, H.Z.; Su, Z.L.; Cvirkaite-Krupovic, V.; Krupovic, M.; Egelman, E.H.; Si, D. DeepTracer-ID: De novo protein identification from cryo-EM maps. Biophys. J. 2022, 121, 2840–2848.
- He, J.; Huang, S.Y. Full-length de novo protein structure determination from cryo-EM maps using deep learning. Bioinformatics 2021, 37, 3480–3490.
- Zhou, X.; Li, Y.; Zhang, C.; Zheng, W.; Zhang, G.; Zhang, Y. Progressive assembly of multi-domain protein structures from cryo-EM density maps. Nat. Comput. Sci. 2022, 2, 265–275.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Lin, Z.; Halil, A.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022.
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353.
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721.
- AF2 Is Here: What’s Behind the Structure Prediction Miracle. Available online: https://www.blopig.com/blog/2021/07/alphafold-2-is-here-whats-behind-the-structure-prediction-miracle (accessed on 19 June 2023).
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444.
- Burley, S.K.; Bhikadiya, C.; Bi, C.X.; Bittrich, S.; Chao, H.Y.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2022, 51, D488–D508.
- Andreeva, L.; David, L.; Rawson, S.; Shen, C.; Pasricha, T.; Pelegrin, P.; Wu, H. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 2021, 184, 6299–6312.
- Noddings, C.M.; Wang, R.Y.; Johnson, J.L.; Agard, D.A. Structure of Hsp90-p23-GR reveals the Hsp90 client-remodelling mechanism. Nature 2022, 601, 465–469.
- Jones, M.L.; Baris, Y.; Taylor, M.R.G.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. EMBO J. 2021, 40, e108819.
- Fontana, P.; Dong, Y.; Pi, X.; Tong, A.B.; Hecksel, C.W.; Wang, L.F.; Fu, T.M.; Bustamante, C.; Wu, H. Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold. Science 2022, 376, eabm9326.
- Mosalaganti, S.; Obarska-Kosinska, A.; Siggel, M.; Taniguchi, R.; Turonova, B.; Zimmerli, C.E.; Buczak, K.; Schmidt, F.H.; Margiotta, E.; Mackmull, M.T.; et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 2022, 376, eabm9506.
- Terwilliger, T.C.; Poon, B.K.; Afonine, P.V.; Schlicksup, C.J.; Croll, T.I.; Millan, C.; Richardson, J.S.; Read, R.J.; Adams, P.D. Improved AlphaFold modeling with implicit experimental information. Nat. Methods 2022, 19, 1376–1382.
- Alshammari, M.; He, J.; Wriggers, W. Refinement of AlphaFold2 Models against Experimental Cryo-EM Density Maps at 4–6Å Resolution. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine, Las Vegas, NV, USA, 6–8 December 2022; pp. 3423–3430.
- Ziemianowicz, D.S.; Kosinski, J. New opportunities in integrative structural modeling. Curr. Opin. Struc. Biol. 2022, 77, 102488.
- Rantos, V.; Karius, K.; Kosinski, J. Integrative structural modeling of macromolecular complexes using Assembline. Nat. Protoc. 2022, 17, 152–176.
- Beckham, K.S.H.; Ritter, C.; Chojnowski, G.; Ziemianowicz, D.S.; Mullapudi, E.; Rettel, M.; Savitski, M.M.; Mortensen, S.A.; Kosinski, J.; Wilmanns, M. Structure of the mycobacterial ESX-5 type VII secretion system pore complex. Sci. Adv. 2021, 7, eabg9923.
- Flacht, L.; Lunelli, M.; Kaszuba, K.; Chen, Z.A.; Reilly, F.J.O.; Rappsilber, J.; Kosinski, J.; Kolbe, M. Integrative structural analysis of the type III secretion system needle complex from Shigella flexneri. Protein Sci. 2023, 32, e4595.
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682.
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877.
- Kawabata, T. Multiple subunit fitting into a low-resolution density map of a macromolecular complex using a gaussian mixture model. Biophys. J. 2008, 95, 4643–4658.
- Kawabata, T. Gaussian-input Gaussian mixture model for representing density maps and atomic models. J. Struct. Biol. 2018, 203, 1–16.
- Sun, M.L.; Liu, M.D.; Shan, H.; Li, K.; Wang, P.; Guo, H.R.; Zhao, Y.Q.; Wang, R.; Tao, Y.W.; Yang, L.Y.; et al. Ring-stacked capsids of white spot syndrome virus and structural transitions with genome ejection. Sci. Adv. 2023, 9, eadd2796.
- Wang, F.B.; Cvirkaite-Krupovic, V.; Krupovic, M.; Egelman, E.H. Archaeal bundling pili of Pyrobaculum calidifontis reveal similarities between archaeal and bacterial biofilms. Proc. Natl. Acad. Sci. USA 2022, 119, e2216660119.
- Allison, T.M.; Degiacomi, M.T.; Marklund, E.G.; Jovine, L.; Elofsson, A.; Benesch, J.L.P.; Landreh, M. Complementing machine learning-based structure predictions with native mass spectrometry. Protein Sci. 2022, 31, e4333.
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Janes, J.; Zalevsky, A.O.; Meszaros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067.
- Si, D.; Chen, J.; Nakamura, A.; Chang, L.; Guan, H. Smart de novo Macromolecular Structure Modeling from Cryo-EM Maps. J. Mol. Biol. 2023, 435, 167967.