1. Introduction
Cryo-
electron microscopy (Cryo-EM
) and single particle analysis (SPA) have become the preferred method for structure determination of biological complexes at near-atomic resolution
[1][2][3][4][5][6][1,2,3,4,5,6]. Such level of detail not only provides crucial insight into molecular mechanisms employed by biological macromolecules, but also may facilitate the design and development of new drugs and therapeutics. The widespread adoption of cryo-EM is primarily credited to recent technological advances, including the introduction of direct electron detectors and improvements in computer hardware and image-processing software
[7] that have enabled routine structure determination at high-resolution (reviewed in
[8]). In contrast to traditional structural biology techniques, such as X-ray crystallography and Nuclear Magnetic Resonance (NMR), cryo-EM requires very small amounts of purified protein in the range of 0.1 to 5 mg/mL
[9]. Additionally, SPA has proven to be a powerful tool for determining the structures of large and dynamic biological complexes that exhibit a range of compositional and conformational heterogeneity
[10][11][12][10,11,12].
The goal of SPA is to calculate a high-resolution 3D structure from the noisy, 2D projections of the specimen produced on the direct electron detector by the beam of high energy electrons. Multiple software packages have been developed to facilitate this process
[13][14][15][16][17][18][19][20][13,14,15,16,17,18,19,20], and a typical SPA workflow is described in
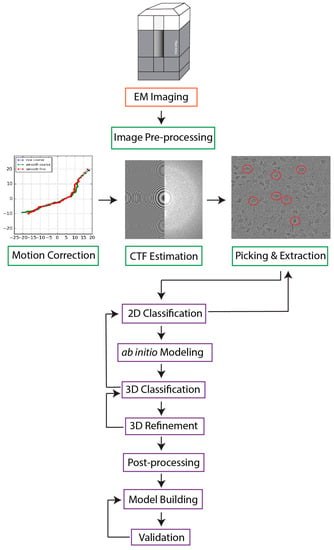
Figure 1. However, several factors complicate image processing: (1) due to advancements in microscope optics, detector technology, and data storage hardware, a single cryo-EM experiment can generate thousands of micrographs with millions of particle images
[21][22][21,22]; (2) biological assemblies are often dynamic and flexible molecular machines that adopt a variety of structural states; (3) cryo-EM micrographs have a low signal-to-noise ratio (SNR), and they are susceptible to artifacts, such as ice contaminations, radiation damage, and preferred orientation of imaged particles, which can obscure underlying structural information. While the application of traditional machine learning (ML) techniques has already enabled the automation of many stages of the workflow
[23][24][25][26][27][28][23,24,25,26,27,28], ab initio reconstruction and atomic model building remain major hurdles in need of robust methodologies to minimize the extensive computational resources and manual user input they currently demand.
Figure 1. A typical SPA workflow. Movies collected with an electron microscope are first motion-corrected. In this step, frames are aligned and averaged to account for beam-induced motion, which increases the SNR of images. The resultant micrographs undergo contrast transfer function (CTF) estimation to calculate the effects of defocus and microscope aberrations. This step is followed by particle picking and extraction, in which particles are selected and extracted from micrographs. The extracted particles are sorted based on orientation into discrete, 2D classes, and the user removes classes containing non-particles, noise, and artifacts. Such filtered particle stacks are used to generate one or more low-resolution ab initio reconstructions that are iteratively refined through 3D classification and 3D refinement to yield final Coulomb potential maps. Given sufficient map resolution and quality, atomic models can be built and validated.
2. Artificial Intelligence-Based Approaches to De Novo Model Building
Early applications of AI methods to model building utilized conventional ML techniques such as k-nearest neighbor (k-NN)
[29][147], k-means clustering
[30][148], and support-vector machines (SVM)
[31][149]. These approaches have been successful in identifying SSEs
[29][31][147,149] and modeling simplified backbone structures
[30][148]. For example, the program RENNSH
[29][147] identifies α-helices in EM maps by representing each voxel as a spherical harmonic descriptor and using a nested k-NN framework to classify α-helix voxels. Another ML approach, SSELearner
[31][149], first learns from maps deposited in the EMDB and then applies an SVM classifier to detect both α-helices and β-sheets in intermediate-resolution density maps. Pathwalking
[30][148] is a de novo model-building approach that uses a combination of the traveling salesman problem and k-means clustering to build a Cα model. Nonetheless, these programs do not guarantee convergence to the correct solution and are not capable of building complete atomic models
[32][33][71,150].
DL has recently driven remarkable advancements in structural biology, including the development of programs for automated de novo atomic model building from cryo-EM density maps (
Table 13). This breakthrough is largely attributed to increased computational power, quality of EM images, and the number of available high-resolution cryo-EM structures for model training
[33][150]. Recently, many CNN-based approaches have been applied not only for automatic assignment of SSEs
[34][35][36][37][50,51,52,53] and backbone chains
[37][38][39][40][53,54,55,56], but also to detect individual amino acids
[40][41][56,57], thus generating complete de novo models
[37][38][39][40][41][53,54,55,56,57] (
Table 13). Below,
rwe
searchers highlight some representative programs utilizing CNN architectures, followed by a description of methods that apply alternative DL frameworks to model building.
AAnchor
[40][56] is the first program with 3D CNN architecture to identify individual amino acids in EM maps. This technique utilizes a classification CNN to locate and label residues, known as anchors, with highest confidence within a defined voxel size
[40][56]. However, AAnchor is currently limited to maps with 3.1 Å resolution or higher and on average, the fraction of anchors detected ranges from 10 to 20% depending on map resolution
[40][56]. Another program, A
2-net
[41][57], uses a CNN framework to identify residues and their poses followed by a Monte Carlo Tree Search (MCTS) strategy to link the 3D coordinate system of amino acids into a complete peptide chain. Compared to its automated counterparts, including Rosetta and others
[42][43][151,152], A
2-net converges to a solution significantly faster. An atomic model for a protein with one thousand amino acids can be derived in minutes
[40][56], compared to several hundred hours in Rosetta
[43][152]. The latest version of A
2-net, CryoNet (
https://cryonet.ai (accessed on 10 May 2023)), has been applied for model building of cryo-EM maps of the human minor spliceosome
[44][153] and the
Acidobacteria homodimeric reaction center bound with cytochrome c
[45][154]. CR-I-TASSER
[38][54] is a fully automated, hybrid method that combines a 3D-CNN to build Cα trace models with multithreading algorithms to identify homologous Protein Data Bank (PDB) templates for guided structure assembly. Nonetheless, the generated CR-I-TASSER model directly relies on the accuracy of the Cα trace prediction, which authors found to decrease with lower resolution maps
[38][54]. Additionally, this technique requires prior segmentation of the experimental map, further limiting its applicability to low-resolution density maps. Although CNNs remain one of the most popular AI architectures for atomic model building and are the most direct DL method for learning features from density maps, they are not without limitations. While CNNs are considered translation-equivariant, meaning translating the input data will result in translated output, they lack rotational invariance, meaning they do not behave consistently for input data of varying orientation. Moreover, the localization of the convolution mechanism narrows the CNN’s receptive field
[46][47][155,156], potentially limiting their ability to capture global features and dependencies within cryo-EM maps.
In addition to standard CNN framework, many de novo model building programs have adopted the U-Net architecture (
Table 1). The U-Net is a type of CNN commonly used to segment or classify pixels in an input image
[48][157]. In the U-Net architecture, an encoder first downsamples the input image and extracts features, and then a decoder network restores the image back to its original dimensions. Through the downsampling and upsampling operations, U-Nets may be more effective at extracting both low-level and high-level features than standard CNNs. Moreover, U-Nets incorporate so-called skip connections that, as the name implies, may skip any given layer in the neural network to provide direct connections between different layers of the network. In the U-Net framework, skip connections enable the direct propagation of feature maps from the encoder to the decoder. This unique architecture preserves fine-grained details and facilitates better information flow throughout the network. Various programs have employed a 3D U-Net framework for SSE identification
[49][50][58,59] and complete de novo model building
[51][52][60,62]. DeepTracer
[51][60] is one of the most popular, fully automated model building programs that utilizes a 3D U-Net architecture to predict locations of SSE elements, backbone atoms, and individual amino acids within EM maps. Evolved from Cascaded-CNN
[37][53], DeepTracer employs a series of four U-Nets to perform distinct tasks: locate amino acid positions, locate the protein backbone, identify SSEs, and identify individual residues
[51][60]. Pfab et al. applied DeepTracer to a set of coronavirus-related cryo-EM maps (
Figure 2), and found that, on average, 84% residues match with the corresponding deposited PDB structures
[51][60]. Furthermore, DeepTracer has been utilized to build a variety of atomic models from EM maps, including the human small subunit processome
[53][158], Chikungunya virus replication complex
[54][159], and human caveolin-1 complex
[55][160]. While DeepTracer demonstrates more accurate Cα prediction than its counterparts
[56][57][58][72,128,161], it is currently only applicable to maps with 5 Å resolution or higher. The latest version, DeepTracer-2.0
[59][61], also has the ability to model nucleic acids.
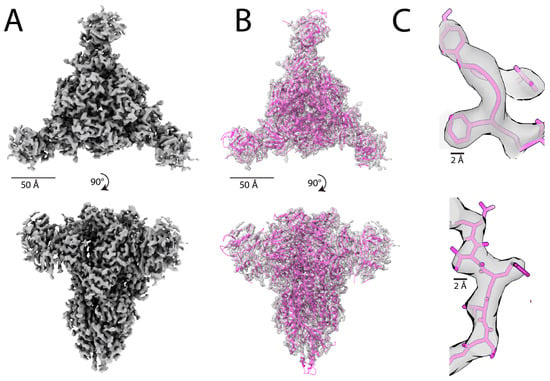
Figure 2. The atomic model built from the cryo-EM map of a feline coronavirus spike protein
[60][162] using DeepTracer
[51][60]. (
A) The 3.3 Å cryo-EM map of a feline coronavirus spike protein (EMDB ID: EMD-9891) that contains 1403 residues. (
B) Density map fitted with the DeepTracer model. The DeepTracer model was built in just 14 min, compared to over 60 h required for model building with Phenix
[51][60]. (
C) Visualization of individual backbone atoms and side chains fitted to the cryo-EM Coulomb map using the molecular model obtained with DeepTracer.
Other DL architectures for de novo model building include the Graph Neural Network (GNN), Recurrent Neural Network (RNN), Residual Neural Network (ResNet), and Long- and Short-Term Memory network (LSTM) (
Table 13). For example, Structure Generator
[39][55] is a fully automated pipeline that utilizes 3D-CNN, GCN, and LSTM algorithms to produce a protein structure based on amino acid identities and locations. Structure Generator
[39][55] first uses a 3D-CNN, named RotamerNet, to locate amino acids and assign their rotameric orientations within the map. Using the Cα coordinates defined by RotamerNet, the GCN generates a contact map and connects Cα positions located within 4 Å. Lastly, the bidirectional LSTM labels the candidate amino acids and ensures the consistency of the assignment with a provided protein sequence to yield a structural model
[39][55]. While Structure Generator has demonstrated high prediction accuracy using simulated datasets
[39][55], it has not yet been applied to experimental maps. Additionally, the program is limited to modeling protein sequences with less than 700 amino acids
[39][55].
DL techniques have provided much needed insight into the automation of atomic model building. Several approaches have demonstrated capabilities to construct full atomic models, but their applicability is generally limited to maps with 5 Å or higher resolution
[51][59][61][62][60,61,64,163]. Furthermore, while several methodologies are able to precisely identify and locate individual atoms, it remains a challenge to construct a full peptide chain without violating geometric and stereochemical restraints
[32][63][65,71]. Different solutions have been proposed to remedy this problem, including the program ModelAngelo
[61][62][64,163] that utilizes sequence data and prior information of protein geometries to refine the protein chain geometry. Another software, CR-I-TASSER, attempts to overcome this limitation by integrating Molecular Dynamics simulations with DL model building
[38][54]. New strategies have also been proposed to integrate DL frameworks with protein structure prediction techniques, such as AlphaFold2, discussed in detail in the following section.
Table 1.
Summary of recently developed AI methods for de novo atomic model building.
| Program |
AI Architecture |
Advantages |
Limitations |
| Emap2Sec [34][50] |
CNN |
Identifies SSEs
Demonstrated on experimental maps
High accuracy for intermediate resolution maps |
Does not place α-helices and β-sheets in detected regions
Voxel-based approach fails for large EM maps |
| Emap2Sec+ [36][52] |
ResNet |
Identifies SSEs and nucleic acids in maps 5–10 Å
Improved accuracy in protein SSE detection compared to Emap2Sec [34][50] |
Voxel-based approach fails for large EM maps |
| CNN-based [35][51] |
CNN |
Identifies SSEs |
Does not place α-helices and β-sheets in detected regions
No results for experimental maps |
| Haruspex [49][58] |
U-Net |
Identifies SSEs and nucleic acids in maps
Applied to experimental and simulated maps |
Only applicable to maps ≤4 Å
False positives for helices, sheets or RNA/DNA
Misclassifies semi-helical elements, β-hairpin turns, polyproline residues as α-helices |
| EMNUSS [50][59] |
U-Net |
Identifies SSEs
Applied to experimental and simulated maps |
Incorrect predictions on atypical density volumes; narrow receptive field |
| AAnchor [40][56] |
CNN |
Identifies amino acids |
Only applicable to maps ≤3.1 Å
Only detects 10–20% amino acids on average |
| A2-Net [41][57] |
3D-CNN, MCTS |
Identifies amino acids
Model with 1000 amino acids can be derived in minutes
Applied to experimental maps
Fully automated |
Cannot identify ligands |
| Structure Generator [39][55] |
3D-CNN,
GCN, LSTM |
Identifies amino acids and rotamer orientation, builds full protein chain |
Limited to protein sequences <700 amino acids
No results for experimental maps |
| CR-I-TASSER [38][54] |
3D-CNN, I-TASSER |
3D-CNN predicts Cα atoms used for I-TASSER; generates full structure |
Cα trace prediction accuracy dependent on resolution
Requires prior map segmentation |
| DeepTracer [51][59][60,61] |
3D U-
Net |
Locates amino acid positions and protein backbone
Identifies SSEs and amino acids
Automated
Lastest version models nucleic acids |
Only applicable to maps ≤5 Å
Only builds atoms for main chains |
| DeepTracer ID [64][63] |
3D U-Net, AlphaFold2 |
Uses DeepTracer to build model and searches against AlphaFold2 library for refinement
Identifies individual proteins in density map; does not require protein sequences to be known a priori
Does not require high accuracy from AlphaFold2 prediction |
Limited to proteins >100 amino acids for succesful AlphaFold2 prediction matches
Only applicable to maps ≤4.2 Å |
| EMBUILD [52][62] |
U-Net, AlphaFold2 |
Constructs main chain map; fits AlphaFold2 predicted chains into the map |
Only builds atoms for main chains |
| ModelAngelo [61][62][64,163] |
CNN, GNN |
Builds complete atomic model
Better RMSD and sequence prediction results than DeepTracer |
Only applicable to maps ≤3.5 Å
Requires protein sequences are known |
| DeepMM [65][164] |
CNN |
Predicts Cα positons; identifies SSEs and amino acids
Applied to experimental maps |
Cannot model nucleic acids
Residue matching accuracy dependent on map resolution |
| DEMO-EM [66][165] |
ResNET, I-TASSER |
Builds complete atomic model for multi-domain proteins
Only requires protein sequence
Fully automated |
Requires prior map segmentation
Individual domain models calculated without restraints from density data |
The field of protein structure prediction has been transformed by significant developments in DL algorithms, exemplified by AlphaFold2
[67][68], RoseTTaFold
[68][166], ESMFold
[69][167], and I-TASSER-MTD
[70][168]. Given only protein sequences, such programs employ a combination of NNs and novel algorithms to predict atomic 3D structures, which in turn provide complementary data to experimental methods. Of particular note, AlphaFold2
[67][68], a publicly available program developed by Google DeepMind, has shown unprecedented levels of accuracy in predicting atomic models
[67][71][72][68,169,170]. AlphaFold2 outperformed all other prediction methods in the 14th edition of the Critical Assessment of Structure Prediction (CASP), a blind test for structure prediction of experimentally solved structures that have not yet been made publicly available. AlphaFold2 models had a median backbone accuracy of 0.96 Å RMSD, whereas the next best performing method had a median backbone accuracy of 2.80 Å RMSD
[67][68]. Using the amino acid sequence as input, AlphaFold2 first performs a Multiple Sequence Alignment (MSA) and tries to identify proteins with similar structures, known as templates. Based on the templates, AlphaFold2 generates a “pair representation” model, indicating which amino acids are likely to be in contact with each other
[73][171]. The program then employs a transformer NN, called the Evoformer, to refine, exchange, and extract information from the MSA and pair representation. The extracted information is used by the structure module, a second NN, to construct an atomic model
[67][68]. The AlphaFold Protein Structure Database
[74][172], hosted at the European Bioinformatics Institute–European Molecular Biology Laboratory (EMBL-EBI), provides access to over 200 million AlphaFold2 models, covering the majority of sequence entries in UniProt. Furthermore, computed models using AlphaFold2 as well as other prediction methods are available through ModelArchive (modelarchive.org). As of 2022, over one million computed models were also accessible through the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB)
[75][173].
Predicted structures can serve as starting models for deriving atomic coordinates from cryo-EM maps
[76][77][78][79][80][174,175,176,177,178]. In addition, several methods have been proposed to dock and fit predicted structures into density maps
[52][81][82][62,66,67]. In cases of multi-domain and multi-component complexes, one approach is to first use AI-prediction tools to generate structures of sub-complexes and then assemble them into higher-order structures
[70][83][84][168,179,180]. For example, the protein structure prediction and modeling tool I-TASSER-MTD
[70][168] predicts structures of individual domains and then uses the EM density to guide the construction of a complete atomic model. Assembline
[84][180] is another software package that offers an integrative pipeline for model building that satisfies the constraints of predicted structures and cryo-EM maps. Assembline has been used to assemble full atomic structures of multiple protein complexes
[80][85][86][178,181,182], including the human nuclear core complex
[80][178]. In this
res
archtudy, Mosalaganti et al. fit models of individual subunits and sub-complexes generated by AlphaFold2
[67][68] and ColabFold
[87][183], respectively, into the density map, followed by structure assembly with Assembline
[84][180]. Notably, the model of the N-terminal domain of NUP358 predicted with AlphaFold2 was in better agreement with the cryo-EM map than atomic coordinates from previously determined X-ray structures
[80][178]. Furthermore, the predicted models not only showed the same subunit interactions previously reported by the crystal structures, but also revealed new interactions
[80][178].
Another approach is to combine structure prediction tools with DL-based model building techniques to aid in the construction and refinement of atomic models
[52][61][63][64][66][81][82][62,63,64,65,66,67,165]. The program EMBUILD
[52][62] utilizes a U-Net to first generate a so-called main-chain probability map including potential solutions and probabilities reflecting the agreement of the main chain assignments with experimental densities. AlphaFold2
[67][68] predicted models of individual chains are then fit into the map, and a scoring function is used to measure how well the fitted chain matches the main-chain probability map. The final model is the combination of fitted chains that yield the highest scoring function among different combinations of fitted positions
[52][62]. He et al.
[52][62] applied EMBUILD to 47 intermediate-resolution maps and built models with an average template modeling score (TM-score) of 0.909 and RMSD of 2.85 Å, outperforming other state-of-the-art methods, including phenix.dock_in_map
[88][184] and gmfit
[89][90][185,186]. Another program, DEMO-EM
[66][165], utilizes DL-based inter-domain distance maps to facilitate the fully automated assembly and refinement of individual domains into full-length structures. When applied to a benchmark set of multi-domain proteins with medium- to low-resolution maps, DEMO-EM generated models with correct inter-domain orientations in 97% of cases, outperforming MDFF and Rosetta
[66][165]. Nonetheless, DEMO-EM, along with the other programs described in this section, utilize models generated for each subunit independently without taking into consideration the conformational changes that may occur upon subunit binding
[61][64].
Other algorithms leverage structure prediction tools to identify proteins within cryo-EM maps. With advancements in image processing techniques, cryo-EM can now be applied directly to cellular extracts. However, determining the specific proteins within the resulting map often requires the application of other complementary techniques, such as tandem Mass Spectrometry. The program DeepTracer-ID
[64][63] combines DeepTracer
[51][59][60,61] and AlphaFold2 to directly identify the proteins present in a cryo-EM map. DeepTracer-ID first utilizes DeepTracer, as described above, to build an atomic structure, and then uses this model to search the AlphaFold2 library containing all predicted structures from any given organism. Identified structures are used to iteratively refine the atomic model. In a blind test of 13 experimental maps, DeepTracer-ID identified the correct proteins as top candidates
[64][63]. DeepTracer-ID has also been successfully used for protein identification in cryo-EM maps of a white spot syndrome virus capsid
[91][187] and archaeal surface filaments
[92][188]. Nonetheless, this approach is only applicable for maps with 4.2 Å or higher resolution. In addition, accurate sequence-based alignment of the DeepTracer model and AlphaFold2 predicted structure requires proteins with a sequence longer than 100 amino acids
[64][63]. Similarly, the program DeepProLigand
[63][65] predicts protein ligand interactions by employing DeepTracer and using other known protein-ligand structures available in AlphaFold2 library or available through the RCSB PDB.
While integrative approaches described in this section represent a promising tool for atomic model building, AI structure prediction methodologies suffer from several limitations. Assessing the quality of predicted AlphaFold2 structures is a nontrivial task. Although quality metrics accompany each AlphaFold2 model, these scores are also predictions that may contain errors
[93][189]. Thus, manual inspection is still required to evaluate the model in the context of the experimental map, as even high-confidence predictions can be modeled incorrectly
[94][190]. Furthermore, as AlphaFold2 relies on experimentally determined structures deposited in the PDB, the algorithm may produce incorrect folds when there is a lack of homological structures
[95][70]. Further developments are needed to improve the prediction accuracy of mutated residues, regions involved in ligand binding, and dynamic interactions
[93][94][189,190].