Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Giuseppe Murdaca.
Osteoarthritis (OA) is a multifactorial disease in which genetics, aging, obesity, and trauma are well-known risk factors. It is the most prevalent joint disease and the largest disability problem worldwide. Recent fFindings have described the role of damage-associated molecular patterns (DAMPs) in the course of the disease. In particular, alarmins such as HMGB1, IL-33, and S100B, appear implicated in enhancing articular inflammation and favouring a catabolic switch in OA chondrocytes.
- osteoarthritis
- alarmins
- DAMPs
- cytokines
- HMGB1
- S100B
- IL-33
1. Introduction
Osteoarthritis (OA) is the most common subtype of arthritis affecting older people [1,2,3][1][2][3]; it is a chronic and degenerative joint disorder characterised by hyaline cartilage damage, subchondral bone remodelling, chondrophyte and osteophyte formation, and synovitis [4,5,6,7][4][5][6][7]. Different risk factors concur in the pathogenesis and the most common are aging, obesity, trauma, female sex, and genetics [8,9,10,11][8][9][10][11]. The first event in the course of the disease is hyaline cartilage degeneration. Cartilage is an avascular and nerveless tissue formed by chondrocytes, the only cell type, and extracellular matrix, secreted by chondrocytes themselves and high in proteoglycans and collagen. Chondrocytes are implicated in cartilage homeostasis; indeed, any insult affecting them, such as mechanical trauma, abuse, and/or repetitive loads, or hypoxia, results in alterations of the extracellular matrix [12]. After a joint injury occurs, a significant quantity of chondrocytes die along the surface of cartilage, and adjacent cells are driven by different mediators to switch into catabolic molecular activities, so the excess of degradative over reparative processes results in gradual loss of articular cartilage and joint space narrowing [13,14,15][13][14][15].
Despite the chronic and degenerative definition of OA, the pathogenesis is driven by different inflammatory mediators, such as cytokines, chemokines, damage-associated molecular patterns (DAMPs), matrix proteinases (MMPs and ADAMTS), and eicosanoids [15,16,17][15][16][17]. Tumor necrosis factor α (TNF-α) and mostly interleukin 1β (IL-1β) are involved in catabolic enzymes, nitric oxide (NO), and prostaglandins synthesis and in suppressing extracellular matrix components production by chondrocytes [14,16,18][14][16][18].
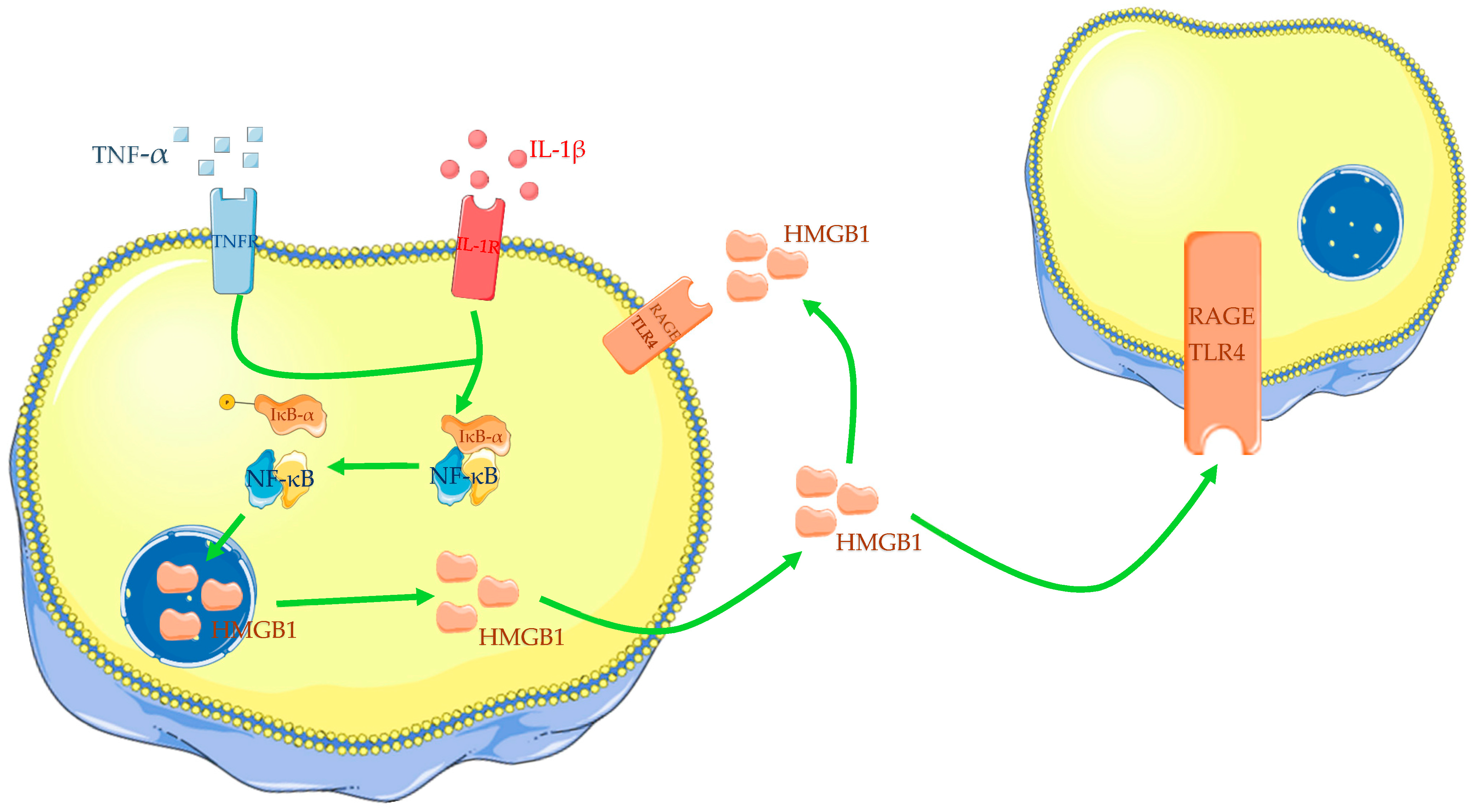
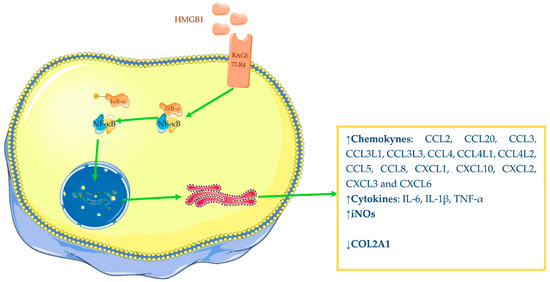 Figure 2. Pro-inflammatory power of HMGB1: increased synthesis of chemokine, cytokines, iNOS and decreased synthesis of COL2A1. The Figure was drawn using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. It was partly modified and adapted with tests.
However, it was largely demonstrated that alarmins do not act alone but in a synergic way with other factors: in particular, HMGB1 increases the IL-1β-induced synthesis of matrix metalloproteinases (MMPs), a disintegrin and metalloproteinse (ADAM), and iNOS by normal human chondrocytes; FN-f shows similar results but with a minor synergic effect on IL-1β stimulated chondrocytes [69][63].
Finally, it is important to underline that OA chondrocytes have shown increased expression of RAGE and TLR4, which are HMGB1 receptors [60,64][54][58]. Apoptosis of chondrocytes results in increased secretion of HMGB1 while senescence induces the accumulation of advanced glycation end products (AGEs); therefore, it is possible to assume that the number of AGEs and HMGB1 in cartilage may favour the interaction with RAGE, thus increasing the secretion of pro-inflammatory cytokines and contributing to cartilage degeneration, further confirming its role in OA [62][56].
It was described that HMGB1 expression is also augmented in OA synovial membrane compared with the normal membrane [67,70,71][61][64][65]. The works of Garcìa-Arnandis et al. and Ke and colleagues have shown that the number of HMGB1-positive OA synoviocytes was higher than that in controls, and HMGB1 positivity was found mostly in the cytoplasm and in the extracellular medium rather than in the nuclei; however, the results of the first paper have not reached statistical significance [71,72][65][66]. Sun and colleagues have reported similar evidence: they have found augmented messenger RNA (mRNA) and protein expressions of HMGB1 and RAGE by synovial tissues from knee OA patients, compared with the control group. HMGB1 positivity was predominantly cytoplasmatic; additionally, the authors demonstrated that HMGB1 and RAGE levels were positively correlated with the X-ray grade of the disease [70][64]. HMGB1 was also found to increase, same as mRNA and protein expressions, in synovial samples from OA temporomandibular joint compared with patients with a condylar fracture [73][67].
HMGB1 seems to act on synoviocytes in the same way as on chondrocytes. In fact, different works have confirmed the inflammation-enhancing power of HMGB1 on synovial cells. It is widely described that this alarmin could increase interleukins, chemokines, and MMPs productions by OA synoviocytes, acting in complex with other pro-inflammatory mediators, such as LPS, IL-1α, and IL-1β. Specifically, the HMGB1-IL-1β combination induces a higher cytokines production than the HMGB1-LPS complex.
HMGB1 has been shown to significantly increase MMP-13, ADAMTS-5, IL-1β, and IL-6 synthesis in human synovial fibroblasts from OA temporomandibular joint (TMJ) through NF-κB signalling [76][68]. It was shown that OA human chondrocytes, stimulated by recombinant dsHMGB1, increased synthesis of NF-κB1 (NF-κB p105 subunit) and NF-κB2 (NF-κB p100 subunit) mRNA, two NF-κB light chain enhancers [63][57].
Jiang and colleagues have demonstrated that HMGB1 augmented expression and translocation from nucleus to cytoplasm in human chondrocytes incubated with IL-1β is due to bromodomain containing 4 (BRD4) increased expression in cartilage from OA-affected patients, compared with non-OA-affected patients. Additionally, BRD4 expression was positively correlated with histological OARSI score, and this finding may partially explain the gradually increased expression of HMGB1 with OA progression. This protein belongs to the bromo and extra-terminal (BET) domain family and plays an epigenetic regulatory role because of its capacity to bind acetylated lysine residues of histone tails. In fact, researchers have postulated and reported that BRD4 acts by binding the HMGB1 promoter and upstream non-promoter region, inducing gene transcription.
MCM3AP antisense RNA 1 (MCM3AP-AS1) is a long noncoding RNA primarily described in hepatocellular carcinoma. Gao and colleagues have reported increased expression of this RNA in OA patients compared with controls. They have demonstrated that MCM3AP-AS1 acts by binding miR-142-3p, a miRNA that may improve OA by targeting HMGB1, thus reducing apoptosis and inflammation.
MiR-140-5p, such as miR-142-3p, confirmed improved effects on OA, reducing apoptosis, pro-inflammatory factors, and matrix metalloproteinases synthesis in IL-1β-induced chondrocytes. It acts by targeting the 3′-UTR of HMGB1, and low levels of miR-140-5p correspond to the high expression of HMGB1 found in OA tissues and IL-1β-induced chondrocytes. Wang et al. have reported that miR-140-5p interferes with PI3K/AKT, a demonstrated downstream pathway of HMGB1; this miRNA has reduced levels of phosphorylated PI3K and phosphorylated AKT (Ser473), as well as HMGB1, in IL-1β-mediated chondrocytes. Finally, miR-140-5p overexpression has revealed reduced levels of MMP-1, MMP-3, TNF-α, IL-6, and apoptosis rate, and increased cell viability [80][69].
MiR-129-5p is another miRNA with described ability to relieve OA alterations. In vitro tests have shown that it targets 3′-UTR of HMGB1 mRNA, reducing the alarmin release and alleviating inflammatory responses and apoptosis of IL-1β-incubated chondrocytes. In fact, IL-1β-mediated chondrocytes incubated with exosomes poor in miR-129-5p have shown increased HMGB1, TLR4, phosphorylated NF-κB, cyclooxygenase 2 (COX2), iNOS, and MMP13 levels and apoptosis rate, while collagen 2 was downregulated. On the other side, the same cells incubated with exosomes rich in miR-129-5p have revealed inverse results.
Plasmacytoma variant translocation 1 (PVT1) is another long noncoding RNA involved in OA pathogenesis. Meng and colleagues have reported higher PVT1 levels in the serum of OA patients and LPS-stimulated human normal chondrocytes C28/I2 than in the controls. On the other side, the miR-93-5p expression has shown a negative correlation with PVT1, with lower levels in the serum of OA patients and LPS-stimulated C28/I2. The researchers have demonstrated that PVT1 is implicated in OA pathogenesis since PVT1 downregulation, through small interference targeting PVT1 (si-PVT1), relieves cytokines secretion, collagen degradation, and apoptosis rate in LPS-incubated C28/I2.
Figure 2. Pro-inflammatory power of HMGB1: increased synthesis of chemokine, cytokines, iNOS and decreased synthesis of COL2A1. The Figure was drawn using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. It was partly modified and adapted with tests.
However, it was largely demonstrated that alarmins do not act alone but in a synergic way with other factors: in particular, HMGB1 increases the IL-1β-induced synthesis of matrix metalloproteinases (MMPs), a disintegrin and metalloproteinse (ADAM), and iNOS by normal human chondrocytes; FN-f shows similar results but with a minor synergic effect on IL-1β stimulated chondrocytes [69][63].
Finally, it is important to underline that OA chondrocytes have shown increased expression of RAGE and TLR4, which are HMGB1 receptors [60,64][54][58]. Apoptosis of chondrocytes results in increased secretion of HMGB1 while senescence induces the accumulation of advanced glycation end products (AGEs); therefore, it is possible to assume that the number of AGEs and HMGB1 in cartilage may favour the interaction with RAGE, thus increasing the secretion of pro-inflammatory cytokines and contributing to cartilage degeneration, further confirming its role in OA [62][56].
It was described that HMGB1 expression is also augmented in OA synovial membrane compared with the normal membrane [67,70,71][61][64][65]. The works of Garcìa-Arnandis et al. and Ke and colleagues have shown that the number of HMGB1-positive OA synoviocytes was higher than that in controls, and HMGB1 positivity was found mostly in the cytoplasm and in the extracellular medium rather than in the nuclei; however, the results of the first paper have not reached statistical significance [71,72][65][66]. Sun and colleagues have reported similar evidence: they have found augmented messenger RNA (mRNA) and protein expressions of HMGB1 and RAGE by synovial tissues from knee OA patients, compared with the control group. HMGB1 positivity was predominantly cytoplasmatic; additionally, the authors demonstrated that HMGB1 and RAGE levels were positively correlated with the X-ray grade of the disease [70][64]. HMGB1 was also found to increase, same as mRNA and protein expressions, in synovial samples from OA temporomandibular joint compared with patients with a condylar fracture [73][67].
HMGB1 seems to act on synoviocytes in the same way as on chondrocytes. In fact, different works have confirmed the inflammation-enhancing power of HMGB1 on synovial cells. It is widely described that this alarmin could increase interleukins, chemokines, and MMPs productions by OA synoviocytes, acting in complex with other pro-inflammatory mediators, such as LPS, IL-1α, and IL-1β. Specifically, the HMGB1-IL-1β combination induces a higher cytokines production than the HMGB1-LPS complex.
HMGB1 has been shown to significantly increase MMP-13, ADAMTS-5, IL-1β, and IL-6 synthesis in human synovial fibroblasts from OA temporomandibular joint (TMJ) through NF-κB signalling [76][68]. It was shown that OA human chondrocytes, stimulated by recombinant dsHMGB1, increased synthesis of NF-κB1 (NF-κB p105 subunit) and NF-κB2 (NF-κB p100 subunit) mRNA, two NF-κB light chain enhancers [63][57].
Jiang and colleagues have demonstrated that HMGB1 augmented expression and translocation from nucleus to cytoplasm in human chondrocytes incubated with IL-1β is due to bromodomain containing 4 (BRD4) increased expression in cartilage from OA-affected patients, compared with non-OA-affected patients. Additionally, BRD4 expression was positively correlated with histological OARSI score, and this finding may partially explain the gradually increased expression of HMGB1 with OA progression. This protein belongs to the bromo and extra-terminal (BET) domain family and plays an epigenetic regulatory role because of its capacity to bind acetylated lysine residues of histone tails. In fact, researchers have postulated and reported that BRD4 acts by binding the HMGB1 promoter and upstream non-promoter region, inducing gene transcription.
MCM3AP antisense RNA 1 (MCM3AP-AS1) is a long noncoding RNA primarily described in hepatocellular carcinoma. Gao and colleagues have reported increased expression of this RNA in OA patients compared with controls. They have demonstrated that MCM3AP-AS1 acts by binding miR-142-3p, a miRNA that may improve OA by targeting HMGB1, thus reducing apoptosis and inflammation.
MiR-140-5p, such as miR-142-3p, confirmed improved effects on OA, reducing apoptosis, pro-inflammatory factors, and matrix metalloproteinases synthesis in IL-1β-induced chondrocytes. It acts by targeting the 3′-UTR of HMGB1, and low levels of miR-140-5p correspond to the high expression of HMGB1 found in OA tissues and IL-1β-induced chondrocytes. Wang et al. have reported that miR-140-5p interferes with PI3K/AKT, a demonstrated downstream pathway of HMGB1; this miRNA has reduced levels of phosphorylated PI3K and phosphorylated AKT (Ser473), as well as HMGB1, in IL-1β-mediated chondrocytes. Finally, miR-140-5p overexpression has revealed reduced levels of MMP-1, MMP-3, TNF-α, IL-6, and apoptosis rate, and increased cell viability [80][69].
MiR-129-5p is another miRNA with described ability to relieve OA alterations. In vitro tests have shown that it targets 3′-UTR of HMGB1 mRNA, reducing the alarmin release and alleviating inflammatory responses and apoptosis of IL-1β-incubated chondrocytes. In fact, IL-1β-mediated chondrocytes incubated with exosomes poor in miR-129-5p have shown increased HMGB1, TLR4, phosphorylated NF-κB, cyclooxygenase 2 (COX2), iNOS, and MMP13 levels and apoptosis rate, while collagen 2 was downregulated. On the other side, the same cells incubated with exosomes rich in miR-129-5p have revealed inverse results.
Plasmacytoma variant translocation 1 (PVT1) is another long noncoding RNA involved in OA pathogenesis. Meng and colleagues have reported higher PVT1 levels in the serum of OA patients and LPS-stimulated human normal chondrocytes C28/I2 than in the controls. On the other side, the miR-93-5p expression has shown a negative correlation with PVT1, with lower levels in the serum of OA patients and LPS-stimulated C28/I2. The researchers have demonstrated that PVT1 is implicated in OA pathogenesis since PVT1 downregulation, through small interference targeting PVT1 (si-PVT1), relieves cytokines secretion, collagen degradation, and apoptosis rate in LPS-incubated C28/I2.
2. Alarmins: HMGB1, IL-33, S100B
Alarmins are a group of mediators that play a role in the innate inflammatory response. They are so named because they are usually released by cells from injured tissues, and thus are also known as damage-associated molecular patterns (DAMPs). In particular, alarmins are endogenous DAMPs, so it is important to distinguish them from exogenous DAMPs, derived from pathogens, the so-called pathogen-associated molecular patterns (PAMPs) [20,21,22][19][20][21].2.1. HMGB1
High mobility group box 1 (HMGB1) is a nuclear nonhistone DNA binding protein, so named for its rapid mobility on gel electrophoresis [23][22]. HMGB1 belongs to the high mobility group proteins (HMGs), which are divided into three families: HMGA, HMGB, and HMGN, respectively, distinguished by the AT-hook domain, two boxes of DNA-binding domains, and a nucleosomal binding domain [24,25,26][23][24][25]. The HMGB family includes four proteins: HMGB1, HMGB2, HMGB3, and HMBG4. HMGB1 has three different domains: box A and box B, two DNA-binding motifs, and acid C-terminal tails [23,27,28][22][26][27]. Box B is the active cytokine domain while box A limits the function of the alarmin [27][26]. HMGB1 can be actively or passively released outside the cell; inflammatory stimulations mediate the translocation from the nucleus to the cytoplasm and then to the extracellular medium, while necrosis and apoptosis are passive modalities of HMGB1 cell release [29,30,31,32,33][28][29][30][31][32]. It has different functions depending on its localisation: in the nucleus, HMGB1 promotes nucleosomal stability and supports the bond between transcriptional factors and their DNA target; when released in the extracellular milieu, it chemo-attracts osteoblasts, osteoclasts, and endothelial cells during endochondral ossifications and promotes transendothelial migration of monocytes and amplification of inflammatory response [23,30,34,35][22][29][33][34]. HMGB1 interacts with multiple receptors: Toll-like receptors (TLR2 and TLR4), C-X-C motif receptor 4 (CXCR4), and receptors for advanced glycation end products (RAGEs), leading to innate immune activation and inflammation amplification. In particular, HMGB1 interactions depend on the status of the three conserved cysteine domains: disulphide HMGB1 (dsHMGB1) interacts with TLR4, inducing cytokines production and inflammation in vitro; all-thiol HMGB1 (atHMGB1) binds to CXCR4 and RAGE, which are involved in chemotaxis and cell recruitment [30,36,37,38,39][29][35][36][37][38]. Another form is oxidised HMGB1, which is produced by the oxidation of all three cysteine residues through the continuous production of reactive oxygen species (ROS), and it appears not to generate any inflammatory response or chemo-attractive function but is involved in inflammation resolution [40][39]. Finally, hyperacetylation of lysine residues in the nuclear localisation sequences (NLSs) is necessary to determine the cytoplasmatic accumulation of HMGB1, as well as HMGB1 pro-inflammatory power acquisition [30,41][29][40]. The pro-inflammatory role of HMGB1 is also mediated by its capacity to form complexes with other agents, such as lipopolysaccharide (LPS), IL-1β, C-X-C motif ligand 12 (CXCL12), interferon γ (INF-γ), and CpG-DNA (synthetic DNA molecules formed by a single strand containing a cytosine followed by a guanine) [42,43,44,45][41][42][43][44]. HMGB1 is able to stimulate different types of cells, such as chondrocytes, monocytes, and lymphocytes. In particular, its pro-inflammatory power depends on cell type and doses. For example, HMGB1-stimulated peripheral blood monocytes release TNF-α, IL-1α, IL-1β, IL-6, IL-8, macrophage inflammatory protein 1 α and β (MIP-1α and MIP-1β) [46][45].2.2. IL-33
IL-33 belongs to the IL-1 cytokines family, which includes IL-1α, IL-1β, IL-1 receptor antagonist, and IL-18 [47,48][46][47]. It is an alarmin released by apoptotic and necrotic cells, epithelial and endothelial stressed cells [49,50][48][49]. It is synthesised as pro-IL-33 and then released in the extracellular medium after cleavage. This interleukin binds various receptors and activates different intracellular signalling. In particular, IL-33 induces NF-κB activation by binding the T1/ST2 (suppressor of tumorigenity) receptor (ST2L), which belongs to the Toll-like/IL-1 receptor family; furthermore, IL-33 enhances other TLRs pathways, especially TLR2 and TLR4. Additionally, the ST2 receptor has three isoforms produced by the same transcript through differential splicing. ST2L is the transmembrane long form expressed by T helper 2 cells (TH2) and mast cells, which mediates the effect of IL-33 in these cell subtypes. Soluble ST2 (sST2) acts as a decoy receptor by reducing IL-33 interactions with other forms of ST2 receptors, attenuating inflammatory response [51][50]. ST2V is expressed in the stomach, small and large intestine, and spleen [52][51].2.3. S100B
S100B is a calcium-binding protein belonging to the S100 family formed by 24 different members participating as intracellular or extracellular regulators. This alarmin mediates several processes inside cells, such as apoptosis, cell proliferation, differentiation, metabolism, and Ca2+ homeostasis. It is released in the extracellular medium and acts as a DAMP, activating different receptors, such as RAGE and TLR4. RAGE and TLR4, when activated by S100B, promote their intracellular signalling of extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK-MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [24][23]. S100B enhances inflammatory responses in lymphocytes, macrophages, cardiac cells, and endothelial and vascular smooth muscle cells [58,59][52][53].3. The Role of Alarmins in Osteoarthritis Pathogenesis
3.1. Role of HMGB1 in Osteoarthritis
In the last years, increasing attention has been directed to the role of HMGB1 in OA initiation and progression. Different papers have confirmed that OA chondrocytes show an augmented expression of HMGB1 compared with normal cartilage [60,61,62,63][54][55][56][57]. Amin and colleagues have reported an increased expression of HMGB1 in the deep layers of OA articular cartilage compared with normal cartilage [63][57]. Additionally, it has been found that HMGB1 levels in the tidemark with subchondral bone are positively correlated with the histopathological grade of OA [60,61][54][55]. HMGB1 expression in OA cartilage is prevalent in the cytoplasm and extracellular medium rather than in normal cells, whose HMGB1 positivity is predominantly nucleosomal [60,61,63,64][54][55][57][58]. HMGB1 is accumulated from necrotic or apoptotic cells and is actively released by OA chondrocytes [60,63,65][54][57][59]. DAMPs, derived from injured cartilage, can induce translocation of HMGB1 from the nucleus to the cytoplasm and then in the extracellular space. Hwang and colleagues have demonstrated that 29-kDa amino-terminal fibronectin fragment (29-kDa FN-f), a DAMP derived from the extracellular matrix and found in synovial fluid of OA patients, increases the extracellular release of HMGB1 by OA chondrocytes. Different papers have also described the influence of some cytokines on HMGB1 expression in OA chondrocytes. In vitro, tests have confirmed that OA chondrocytes, incubated with IL-1β or TNF-α, increase HMGB1 expression and nucleus to cytoplasm translocation (Figure 1) [60,61,66,67][54][55][60][61]. Additionally, IL-1β appears to reduce chondrocyte viability in a dose-dependent manner [67][61]. For all these reasons, many scholars incubate normal or OA chondrocytes with IL-1β or LPS to reproduce in vitro models of OA and perform tests.Figure 1. Cytokines, such as IL-1β and TNF-α, induce translocation of HMGB1 from the nucleus to the cytoplasm and then into the extracellular medium. HMGB1 could act in autocrine and paracrine ways. The Figure was drawn using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. It was partly modified and adapted to the tests.
HMGB1 has demonstrated a pro-inflammatory role in OA—in particular, it is able to induce augmented expression of different mediators. Several studies have reported that chondrocytes from OA-affected patients, incubated with recombinant dsHMGB1, were induced to increase their chemokines, interleukins, and inducible nitric oxide synthase (iNOs) productions, while anabolic synthesis of collagen type II alpha 1 chain (COL2A1) was decreased [63,68][57][62]. Terada and colleagues have also shown that HMGB1 is able to induce OA chondrocyte augmented release of IL-1β and TNF-α (Figure 2) [60][54]. Therefore, available evidence underlines the interrelationship between IL-1β and HMGB1 since in vitro studies have demonstrated the ability of one to increase the expression of the other.
3.2. Role of IL-33 in Osteoarthritis
IL-33 is another DAMP that seems to have a role in OA pathogenesis. It has been reported that OA chondrocytes show an augmented quantity of IL-33 mRNA, in association with augmented levels of IL-37 and other interleukins, expression of receptors, and MMPs, compared with normal human chondrocytes [89,90,91][70][71][72]. Additionally, IL-33 protein expression has revealed a greater amount in osteoarthritic cartilage than in normal controls [68,91][62][72]; the same results have been found for IL-37, ST2, TLR2, TLR4, NF-κB, IL-6, TNF-α levels, and numerous degradative enzymes [89,90][70][71]. It is possible to assume that IL-33 may have a role in OA development. Mechanical stress is unanimously considered the most impacting factor in OA onset, and for this reason, mRNA of IL-33, MMP1, and MMP13 has been measured and found to be higher in weight-bearing cartilage compared with non-weight-bearing areas. In order to deeply understand if different pathogenesis could define OA according to the type of joint, Rai and colleagues have performed quantitative reverse transcription polymerase chain reaction (qRT-PCR) and immunofluorescence in the two groups of tissues: hip and knee cartilages. They have evidenced that mRNA and protein levels of IL-33, TLR2, TLR4, NF-κB, MMP2, and MMP9 were higher in the OA knee compared with the OA hip, while IL-37 protein expression was higher in the hip than in the knee cartilage [89][70].
IL-33 has also demonstrated a pro-inflammatory power; in fact, this alarmin, such as rHMGB1 and LPS, has increased mRNA levels of IL-33, TNF-α, IL-6, TLR2, TLR4, MMP2, MMP9, NF-κB, HMGB1, and RAGE in incubated normal human articular chondrocytes (NHAC) and has determined M1 phenotype conversion of mediated macrophages. Differently, IL-37 has revealed an anti-inflammatory effect on stimulated NHAC. In fact, this interleukin has reduced mRNA expression of several inflammatory mediators, receptors, and HMGB1 in treated cells compared with non-treated cells. Finally, IL-37 has interfered with the effect of IL-33 since mRNA levels of TLR2, TLR4, IL-6, TNF-α, NF-κB, MMP2, MMP9, RAGE, and HMGB1 decreased in cells incubated with IL-37 followed by IL-33, LPS or rHMGB1 compared with cells incubated only with IL-33, LPS or HMGB1.
Great attention has been directed to the molecular pathway of IL-33; in particular, Li and colleagues have demonstrated that IL-33 is released by chondrocytes after double strand RNAs (dsRNAs) bind TLR3. The researchers have realised a three-step experiment: first, they incubated normal human chondrocytes with supernatant from healthy and injured cartilage lysates and then they compared normal human chondrocytes with supernatant from injured cartilage lysates, with and without RNase A. In the last step, they compared results from human chondrocytes stimulated with supernatant from injured cartilage lysates, knocking down TLR3 and TLR7 with siRNA. They have reported that chondrocytes stimulated with damaged cartilage lysate increase mRNA and protein expression of IL-33, MMP1, and MMP13 and reduce collagen II production. The introduction of RNase A in the lysate reduces this effect, and knocking down TLR3 in stimulated chondrocytes is more effective in reducing IL-33, MMP1, and MMP13 levels and increasing collagen type II expression than silencing TLR7.
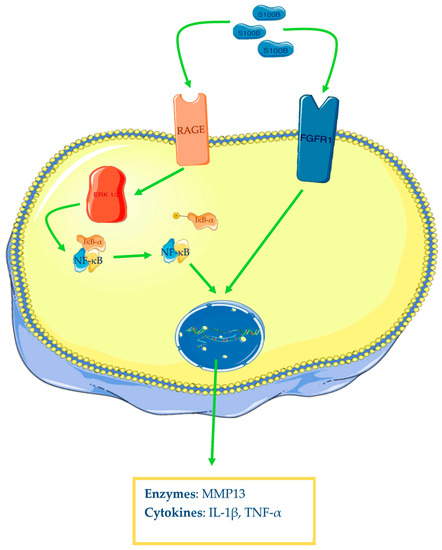
3.3. Role of S100B in Osteoarthritis
S100B appears to be also involved in OA pathogenesis. Zhu and colleagues have reported that OA human cartilage samples show augmented levels of S100B, TNF-α, and IL-1β, but more interesting is the described strong correlation between the levels of the alarmin and the two cytokines. Furthermore, the authors have demonstrated that human synovial fibroblasts from healthy knee patients with S100B overexpression and incubated in vitro with LPS increase their protein expression of TNF-α and IL-1β and mRNA and protein levels of fibroblast growth factor receptor 1 (FGFR1). This has been confirmed by the inverse results found with S100B knockdown through small interfering RNA (siRNA). Finally, the researchers have demonstrated that this alarmin acts through FGFR1 signalling; in fact, FGFR1 knockdown markedly reduced TNF-α and IL-1β levels in the conditioned medium of S100B-overexpressing fibroblasts, incubated with LPS [93][73]. RAGE is an important multiligand receptor whose role in OA has been recently demonstrated. As well as HMGB1, S100B demonstrates binding ability to this receptor. In fact, it has been demonstrated that human chondrocytes incubated with S100B and HMGB1 increase phosphorylated ratios of ERK-1/2 and p65, one of the NF-κB subunits, and protein levels of MMP-13, an enzyme with a key role in OA, for its digestive ability on type II collagen. All these findings are summarised in the Figure 43.Figure 43. S100B and osteoarthritis. After ligand–receptor interaction, the MAP kinases system will be activated (only ERK1/2 is represented in the figure), leading to NF-κB activation and transcription of different genes (cytokines, enzymes) in chondrocytes. The Figure was drawn using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. It was partly modified and adapted with tests.
Osteoarthritic joints show high levels of HMGB1, S100B, and IL-33; particularly, HMGB1 is increased in synovium and synovial fluid, as well as in cartilage. It is possible to assume that a primary injury against cartilage terminates with an initial discharge of HMGB1 that gives a start to a series of molecular events, turning all joint tissues into damaged. In fact, cell death leads to the passive release of HMGB1 in extracellular space, and it may act on adjacent cells, augmenting the expression of cytokines, chemokines, and enzymes and reducing the anabolic expression of collagen. Like in chondrocytes’ necrosis and/or apoptosis, extracellular matrix-derived molecules may induce active translocation of HMGB1 from the nucleus to the cytoplasm and then in the extracellular medium.
Afterwards, HMGB1-induced cytokines release, such as IL-1β and TNF-α, may determine augmented synthesis and release of HMGB1 [60,61,66][54][55][60]. This way, a positive feedback circuit may be generated with the initiation and amplification of sterile inflammation.
It is necessary to underline that OA arises from reiterated traumas, especially in joints with an unbalanced weight distribution. Hence, repeated impacts may set reiterated cell deaths and HMGB1 releases, constantly powering the circuit. These concepts are sustained by observations explaining that HMGB1 levels are correlated with histological grade of OA severity [60,61][54][55].
High levels of HMGB1 in synovial fluid of OA joints have been found positively correlated with the radiological grade of severity (Kellgren-Lawrence system) [84][74] and clinical manifestations [72][66]. For all these reasons, alarmins, specifically HMGB1, may represent a completely new therapeutic target. In vitro, tests have shown that HMGB1 A-box, miR-129-5p, miR-142-3p, miR-140-5p, miR-93-5p, miR-107, chrysin, and glycyrrhizin can counter HMGB1 expression and improve the human osteoarthritic chondrocytes phenotype (rate of apoptosis and catabolic activities) [66,67,77,79,80,81,82,88][60][61][69][75][76][77][78][79].
Nowadays, osteoarthritis is increasingly considered an inflammatory disease with systemic involvement. In a mice model of osteoarthritis, high expression of AGEs and HMGB1 has been found in articular structures and has shown a positive correlation with histological damage in cartilage. Similarly, increased levels of different cytokines, in particular IL-1β, have been revealed in sera of the animals and a positive relationship with joint changes has been reported. Therefore, Kyostio-Moore and colleagues have demonstrated that OA in mice models is a disease with local and systemic inflammatory burden [95][80].
Different papers have demonstrated the beneficial effects of HMGB1 blocking in animal models of OA. Aulin and colleagues have realised three different modalities of intra-articular injections of anti-HMGB1 in mice with OA induced by anterior cruciate ligament transection (ACLT) of the left knee joint. They have effectuated a single administration at the time of surgery (time 0) in the first group, two administrations at 0 and 2 weeks after the surgery in the second group, and two administrations at 2 and 4 weeks after the surgery in the third group.
HMGB1 has also been demonstrated to play a role in the pathogenesis of murine arthritis models. Ostberg and colleagues have reported that nuclear retention of this alarmin through oxaliplatin intra-peritoneal treatment relieves collagen type II-induced arthritis in mice [98][81]. Kokkola and colleagues have illustrated an example of collagen-induced arthritis in mice and rats. The scholars have reported that polyclonal anti-HMGB1 antibodies and HMGB1 A-box intra-peritoneal therapies, reduce clinical disease activity and histologic damage [99][82]. The same results have been obtained by Schierbek and colleagues in their collagen-induced arthritis and spontaneous arthritis in mice with DNase type II and interferon type I receptor deficiencies, treated with intra-peritoneal polyclonal anti-HMGB1 antibodies [100][83].
References
- Lawrence, R.C.; Helmick, C.G.; Arnett, F.C.; Deyo, R.A.; Felson, D.T.; Giannini, E.H.; Heyse, S.P.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998, 41, 778–799.
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part II. Arthritis Rheum. 2008, 58, 26–35.
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387.
- Pelletier, J.-P.; Martel-Pelletier, J.; Abramson, S.B. Osteoarthritis, an inflammatory disease: Potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001, 44, 1237–1247.
- Hill, C.L.; Hunter, D.J.; Niu, J.; Clancy, M.; Guermazi, A.; Genant, H.; Gale, D.; Grainger, A.; Conaghan, P.; Felson, D.T. Synovitis detected on magnetic resonance imaging and its relation to pain and cartilage loss in knee osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1599–1603.
- Smith, M.D.; Triantafillou, S.; Parker, A.; Youssef, P.P.; Coleman, M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol. 1997, 24, 365–371.
- Bobinac, D.; Spanjol, J.; Zoricic, S.; Maric, I. Changes in articular cartilage and subchondral bone histomorphometry in osteoarthritic knee joints in humans. Bone 2003, 32, 284–290.
- Hussain, S.; Neilly, D.; Baliga, S.; Patil, S.; Meek, R. Knee osteoarthritis: A review of management options. Scott. Med. J. 2016, 61, 7–16.
- Messier, S.P. Osteoarthritis of the knee and associated factors of age and obesity: Effects on gait. Med. Sci. Sports Exerc. 1994, 26, 1446–1452.
- Powell, A.; Teichtahl, A.J.; E Wluka, A.; Cicuttini, F.M. Obesity: A preventable risk factor for large joint osteoarthritis which may act through biomechanical factors. Br. J. Sports Med. 2005, 39, 4–5.
- Sacitharan, P.K. Ageing and Osteoarthritis. In Biochemistry and Cell Biology of Ageing: Part II Clinical Science ; Harris, J.R., Korolchuk, V.I., Eds.; Subcellular Biochemistry; Springer: Singapore, 2019; Volume 91, pp. 123–159. Available online: http://link.springer.com/10.1007/978-981-13-3681-2_6 (accessed on 22 April 2023).
- Buckwalter, J.A. Osteoarthritis and articular cartilage use, disuse, and abuse: Experimental studies. J. Rheumatol. 1995, 43, 13–15.
- Smith, R.L.; Trindade, M.C.; Ikenoue, T.; Mohtai, M.; Das, P.; Carter, D.R.; Goodman, S.B.; Schurman, D.J. Effects of shear stress on articular chondrocyte metabolism. Biorheology 2000, 37, 95–107.
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707.
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592.
- Poole, R.; Guilak, F.; Abramson, S.B. Etiopathogenesis of osteoarthritis. Osteoarthr. Diagn. Med./Surg. Manag. 2007, 4, 27–49.
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr. Cartil. 2013, 21, 16–21.
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208.
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045.
- Castiglioni, A.; Canti, V.; Rovere-Querini, P.; Manfredi, A.A. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 2011, 343, 189–199.
- Danieli, M.G.; Antonelli, E.; Piga, M.A.; Claudi, I.; Palmeri, D.; Tonacci, A.; Allegra, A.; Gangemi, S. Alarmins in autoimmune diseases. Autoimmun. Rev. 2022, 21, 103142.
- Štros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1799, 101–113.
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64.
- Cleynen, I.; Van de Ven, W.J. The HMGA proteins: A myriad of functions (Review). Int. J. Oncol. 2008, 32, 289–305. Available online: http://www.spandidos-publications.com/10.3892/ijo.32.2.289 (accessed on 22 April 2023).
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; A Amoscato, A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17.
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol. Ther. 2014, 141, 347–357.
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849.
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195.
- Andersson, U.; Antoine, D.J.; Tracey, K.J. Expression of Concern: The functions of HMGB 1 depend on molecular localization and post-translational modifications. J. Intern. Med. 2014, 276, 420–424.
- Jiang, W.; Bell, C.W.; Pisetsky, D.S. The Relationship between Apoptosis and High-Mobility Group Protein 1 Release from Murine Macrophages Stimulated with Lipopolysaccharide or Polyinosinic-Polycytidylic Acid. J. Immunol. 2007, 178, 6495–6503.
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous Danger Signaling. Mol. Med. 2008, 14, 476–484.
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.-H.; Taxman, D.J.; Duncan, J.A.; Ting, J.P.-Y. NLRP3 (NALP3, Cryopyrin) Facilitates In Vivo Caspase-1 Activation, Necrosis, and HMGB1 Release via Inflammasome-Dependent and -Independent Pathways. J. Immunol. 2009, 183, 2008–2015.
- Rouhiainen, A.; Kuja-Panula, J.; Wilkman, E.; Pakkanen, J.; Stenfors, J.; Tuominen, R.K.; Lepäntalo, M.; Carpén, O.; Parkkinen, J.; Rauvala, H. Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004, 104, 1174–1182.
- Taniguchi, N.; Yoshida, K.; Ito, T.; Tsuda, M.; Mishima, Y.; Furumatsu, T.; Ronfani, L.; Abeyama, K.; Kawahara, K.-I.; Komiya, S.; et al. Stage-Specific Secretion of HMGB1 in Cartilage Regulates Endochondral Ossification. Mol. Cell. Biol. 2007, 27, 5650–5663.
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdés-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1—Dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14.
- Yang, H.; Lundbäck, P.; Ottosson, L.; Erlandsson-Harris, H.; Venereau, E.; Bianchi, M.E.; Al-Abed, Y.; Andersson, U.; Tracey, K.J. Redox modifications of cysteine residues regulate the cytokine activity of HMGB1. Mol. Med. 2021, 27, 1–7.
- Chavakis, E.; Hain, A.; Vinci, M.; Carmona, G.; Bianchi, M.E.; Vajkoczy, P.; Zeiher, A.M.; Chavakis, T.; Dimmeler, S. High-Mobility Group Box 1 Activates Integrin-Dependent Homing of Endothelial Progenitor Cells. Circ. Res. 2007, 100, 204–212.
- Andersson, U.; Yang, H.; Harris, H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin. Ther. Targets 2018, 22, 263–277.
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528.
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873.
- Youn, J.H.; Oh, Y.J.; Kim, E.S.; Choi, J.E.; Shin, J.-S. High Mobility Group Box 1 Protein Binding to Lipopolysaccharide Facilitates Transfer of Lipopolysaccharide to CD14 and Enhances Lipopolysaccharide-Mediated TNF-α Production in Human Monocytes. J. Immunol. 2008, 180, 5067–5074.
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 Develops Enhanced Proinflammatory Activity by Binding to Cytokines. J. Immunol. 2008, 180, 2531–2537.
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563.
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496.
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570.
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490.
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287.
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? Unutmaz D, curatore. PLoS ONE. 2008, 3, e3331.
- Nakae, S.; Morita, H.; Ohno, T.; Arae, K.; Matsumoto, K.; Saito, H. Role of Interleukin-33 in Innate-Type Immune Cells in Allergy. Allergol. Int. 2013, 62, 13–20.
- Vladimir, T.; Sweet, M.J.; Xu, D. T1/ST2—An IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 2004, 15, 87–95.
- Tago, K.; Noda, T.; Hayakawa, M.; Iwahana, H.; Yanagisawa, K.; Yashiro, T.; Tominaga, S.-i. Tissue Distribution and Subcellular Localization of a Variant Form of the Human ST2 Gene Product, ST2V. Biochem. Biophys. Res. Commun. 2001, 285, 1377–1383.
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Tubaro, C.; Bianchi, R.; Giambanco, I.; Donato, R. S100B protein in tissue development, repair and regeneration. World J. Biol. Chem. 2013, 4, 1.
- Sorci, G.; Bianchi, R.; Riuzzi, F.; Tubaro, C.; Arcuri, C.; Giambanco, I.; Donato, R. S100B Protein, a Damage-Associated Molecular Pattern Protein in the Brain and Heart, and Beyond. Cardiovasc. Psychiatry Neurol. 2010, 2010, 1–13.
- Terada, C.; Yoshida, A.; Nasu, Y.; Mori, S.; Tomono, Y.; Tanaka, M.; Takahashi, H.; Nishibori, M.; Ozaki, T.; Nishida, K. Gene expression and localization of high-mobility group box chromosomal protein-1 (HMGB-1)in human osteoarthritic cartilage. Acta Med. Okayama 2011, 65, 369–377.
- Heinola, T.; Kouri, V.-P.; Clarijs, P.; Ciferska, H.; Sukura, A.K.K.; Salo, J.; Konttinen, Y.T. High mobility group box-1 (HMGB-1) in osteoarthritic cartilage. Ann. Rheum. Dis. 2010, 28, 511–518.
- Rosenberg, J.H.; Rai, V.; Dilisio, M.F.; Sekundiak, T.D.; Agrawal, D.K. Increased expression of damage-associated molecular patterns (DAMPs) in osteoarthritis of human knee joint compared to hip joint. Mol. Cell. Biochem. 2017, 436, 59–69.
- Amin, A.R.; Islam, A.B.M.M.K. Genomic Analysis and Differential Expression of HMG and S100A Family in Human Arthritis: Upregulated Expression of Chemokines, IL-8 and Nitric Oxide by HMGB1. DNA Cell Biol. 2014, 33, 550–565.
- Wagner, G.; Lehmann, C.; Bode, C.; Miosge, N.; Schubert, A. High Mobility Group Box 1 Protein in Osteoarthritic Knee Tissue and Chondrogenic Progenitor Cells: An Ex Vivo and In Vitro Study. Cartilage 2019, 12, 484–495.
- Hwang, H.S.; Choi, M.H.; Kim, H.A. 29-kDa FN-f inhibited autophagy through modulating localization of HMGB1 in human articular chondrocytes. BMB Rep. 2018, 51, 508–513.
- Fu, Y.; Lei, J.; Zhuang, Y.; Zhang, K.; Lu, D. Overexpression of HMGB1 A-box reduced IL-1β-induced MMP expression and the production of inflammatory mediators in human chondrocytes. Exp. Cell Res. 2016, 349, 184–190.
- Zhou, S.; Liu, G.; Si, Z.; Yu, L.; Hou, L. Glycyrrhizin, an HMGB1 inhibitor, Suppresses Interleukin-1β-Induced Inflammatory Responses in Chondrocytes from Patients with Osteoarthritis. CARTILAGE 2021, 13 (Suppl. 2), 947S–955S.
- Aulin, C.; Lassacher, T.; Palmblad, K.; Harris, H.E. Early stage blockade of the alarmin HMGB1 reduces cartilage destruction in experimental OA. Osteoarthr. Cartil. 2020, 28, 698–707.
- Ding, L.; Buckwalter, J.A.; Martin, J.A. DAMPs Synergize with Cytokines or Fibronectin Fragment on Inducing Chondrolysis but Lose Effect When Acting Alone. Mediat. Inflamm. 2017, 2017, 1–12.
- Sun, X.-H.; Liu, Y.; Han, Y.; Wang, J. Expression and Significance of High-Mobility Group Protein B1 (HMGB1) and the Receptor for Advanced Glycation End-Product (RAGE) in Knee Osteoarthritis. Experiment 2016, 22, 2105–2112.
- García-Arnandis, I.; Guillén, M.I.; Gomar, F.; Pelletier, J.P.; Martel-Pelletier, J.; Alcaraz, M.J. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1b in osteoarthritic synoviocytes. Arthritis Res. Ther. 2010, 12, R165.
- Ke, X.; Jin, G.; Yang, Y.; Cao, X.; Fang, R.; Feng, X.; Lei, B. Synovial Fluid HMGB-1 Levels are Associated with Osteoarthritis Severity. Clin. Lab. 2015, 61, 809–818.
- Feng, Y.; Fang, W.; Li, C.; Guo, H.; Li, Y.; Long, X. The expression of high-mobility group box protein-1 in temporomandibular joint osteoarthritis with disc perforation. J. Oral Pathol. Med. 2015, 45, 148–152.
- Li, Y.Y.; Feng, Y.P.; Liu, L.; Ke, J.; Long, X. Inhibition of HMGB1 suppresses inflammation and catabolism in temporomandibular joint osteoarthritis via NF-κB signaling pathway. Eur. J. Histochem. 2022, 66, 3357. Available online: https://www.ejh.it/index.php/ejh/article/view/3357 (accessed on 27 April 2023).
- Wang, Y.; Shen, S.; Li, Z.; Li, W.; Weng, X. MIR-140-5p affects chondrocyte proliferation, apoptosis, and inflammation by targeting HMGB1 in osteoarthritis. Inflamm. Res. 2020, 69, 63–73.
- Rai, V.; Dilisio, M.F.; Samadi, F.; Agrawal, D.K. Counteractive Effects of IL-33 and IL-37 on Inflammation in Osteoarthritis. Int. J. Environ. Res. Public Health 2022, 19, 5690.
- Li, C.; Chen, K.; Kang, H.; Yan, Y.; Liu, K.; Guo, C.; Qi, J.; Yang, K.; Wang, F.; Guo, L.; et al. Double-stranded RNA released from damaged articular chondrocytes promotes cartilage degeneration via Toll-like receptor 3-interleukin-33 pathway. Cell Death Dis. 2017, 8, e3165.
- He, Z.; Song, Y.; Yi, Y.; Qiu, F.; Wang, J.; Li, J.; Jin, Q.; Sacitharan, P.K. Blockade of IL-33 signalling attenuates osteoarthritis. Clin. Transl. Immunol. 2020, 9, e1185. Available online: https://onlinelibrary.wiley.com/doi/10.1002/cti2.1187 (accessed on 28 April 2023).
- Zhu, L.; Weng, Z.; Shen, P.; Zhou, J.; Zeng, J.; Weng, F.; Zhang, X.; Yang, H. S100B regulates inflammatory response during osteoarthritis via fibroblast growth factor receptor 1 signaling. Mol. Med. Rep. 2018, 18, 4855–4864. Available online: http://www.spandidos-publications.com/10.3892/mmr.2018.9523 (accessed on 28 April 2023).
- Li, Z.-C.; Cheng, G.-Q.; Hu, K.-Z.; Li, M.-Q.; Zang, W.-P.; Dong, Y.-Q.; Wang, W.-L.; Liu, Z.-D. Correlation of synovial fluid HMGB-1 levels with radiographic severity of knee osteoarthritis. Clin. Invest. Med. 2011, 34, E298.
- Qiu, M.; Liu, D.; Fu, Q. MiR-129-5p shuttled by human synovial mesenchymal stem cell-derived exosomes relieves IL-1β induced osteoarthritis via targeting HMGB1. Life Sci. 2021, 269, 118987.
- Gao, Y.; Zhao, H.; Li, Y. LncRNA MCM3AP-AS1 regulates miR-142-3p/HMGB1 to promote LPS-induced chondrocyte apoptosis. BMC Musculoskelet. Disord. 2019, 20, 605.
- Meng, Y.; Qiu, S.; Sun, L.; Zuo, J. Knockdown of exosome-mediated lnc-PVT1 alleviates lipopolysaccharide-induced osteoarthritis progression by mediating the HMGB1/TLR4/NF-κB pathway via miR-93-5p. Mol. Med. Rep. 2020, 22, 5313–5325.
- Lin, S.S.; Yuan, L.J.; Niu, C.C.; Tu, Y.K.; Yang, C.Y.; Ueng, S.W.N. Hyperbaric oxygen inhibits the HMGB1/RAGE signaling pathway by upregulating Mir-107 expression in human osteoarthritic chondrocytes. Osteoarthr. Cartil. 2019, 27, 1372–1381.
- Zhang, C.; Yu, W.; Huang, C.; Ding, Q.; Liang, C.; Wang, L.; Hou, Z.; Zhang, Z. Chrysin protects human osteoarthritis chondrocytes by inhibiting inflammatory mediator expression via HMGB1 suppression. Mol. Med. Rep. 2018, 19, 1222–1229. Available online: http://www.spandidos-publications.com/10.3892/mmr.2018.9724 (accessed on 27 April 2023).
- Kyostio-Moore, S.; Nambiar, B.; Hutto, E.; Ewing, P.J.; Piraino, S.; Berthelette, P.; Sookdeo, C.; Matthews, G.; Armentano, D. STR/ort mice, a model for spontaneous osteoarthritis, exhibit elevated levels of both local and systemic inflammatory markers. Comp. Med. 2011, 61, 346–355.
- Ostberg, T.; Wahamaa, H.; Palmblad, K.; Ito, N.; Stridh, P.; Shoshan, M.; Lotze, M.T.; Harris, H.E.; Andersson, U. Oxaliplatin retains HMGB1 intranuclearly and ameliorates collagen type II-induced arthritis. Thromb. Haemost. 2008, 10, R1.
- Kokkola, R.; Li, J.; Sundberg, E.; Aveberger, A.-C.; Palmblad, K.; Yang, H.; Tracey, K.J.; Andersson, U.; Harris, H.E. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003, 48, 2052–2058.
- Schierbeck, H.; Lundbäck, P.; Palmblad, K.; Klevenvall, L.; Erlandsson-Harris, H.; Andersson, U.; Ottosson, L. Monoclonal Anti-HMGB1 (High Mobility Group Box Chromosomal Protein 1) Antibody Protection in Two Experimental Arthritis Models. Mol. Med. 2011, 17, 1039–1044.
More