1. Introduction
1.1. Glioblastoma Multiforme
Glioblastoma multiforme (GB) is an aggressive cancer with poor prognosis as it is one of the most difficult cancers to treat
[1]. Numerous studies speculate that GB cancer progression may result from a complex multifactorial process implicating different cell types, i.e., astrocytes, neural stem cells, and oligodendrocyte progenitor cells
[1][2][3][4][5][1–5]. Traits present in the tumor microenvironment (TME) or peritumoral niche have been considered very useful to understand the biology of GB progression as they contribute to tumor immune evasion and support tumor growth
[6][7][8,11]. Different immune populations of microglia and macrophages, known as tumor-associated macrophages (TAMs), are regularly found in the peritumoral niche
[7][8][11,12]. Thus, GB progression is strongly afected b
y cells present in the peritumoral niche, and studies involving peritumoral cells have become vitally important (
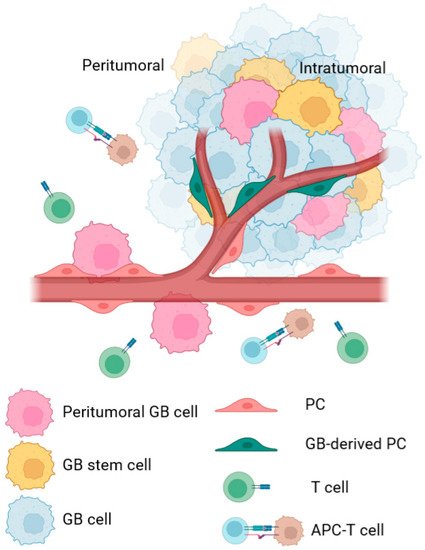
Figure 1). One of these cell types is the pericyte (PC), which resides in the perivascular space of the brain microvessels where GB infiltrates and progression
[9][10][16,17].
Figure 1. Schematic representation of cell type differences in peritumoral and intratumoral areas of GB cancer. In the peritumoral area, peripheral GB cells interact with peritumoral PCs from pre-existent blood vessels and promote several changes in their phenotype. These changes facilitate processes such as co-optation or peritumoral immune evasion, which help with tumor survival, tumor cell infiltration, and tumor spreading in the brain. In the intratumoral area, most studies identify PCs as cells derived from the tumor, which includes not only regular PCs, but also PCs derived from GSC. These PCs are defined to help in angiogenesis or immune evasion of the tumor. In both areas, the cellular changes are intended to promote tumor survival and growth.
1.2. Chaperone-Mediated AutophagyA in Cancer
Chaperone-mediated autophagy CMA is a selective autophagy pathway that degrades specific proteins in lysosomes. Proteins are directly recognized one-by-one by the chaperone heat shock cognate 70 (Hsc70), although other chaperones can be found in the Hsc70-substrate protein complex. Hsc70 recognizes specific KFERQ or KFERQ-like motifs present in the substrate protein, which can be generated via post-transcriptional modifications [11][12][20,21]. The only limiting factor for CMA to proceed is the specific receptor located at the lysosomal membrane, the lysosomal-associated membrane protein 2A (LAMP-2A), which is a splice variant of the lamp2 gene. During CMA, the substrate protein binds to Hsc70 and then to LAMP-2A at the lysosomal membrane to be unfolded, translocated, and degraded within the lysosome with the participation of another chaperone Hsc70, which resides within the lysosome lumen [12][13][14][21–23].
Transformed cells abnormally upregulate CMA activity, which degrades different tumor cell survival- and proliferation-inhibitory proteins [15][16][17][32,34,36]. In addition to upregulation of LAMP-2A expression and, therefore, increased CMA activity in tumor cells, GB-mediated upregulation of CMA in PCs is also required for tumor growth [10][17].
1.3. Peritumoral Pericyte
1.3. Peritumoral PC
PCs are perivascular stromal cells found in the abluminal wall of venules and can be found in different tissues
[18][19][40,41]. PCs derive from different embryonic sources and their phenotype depends on the type of tissue and environment in which they are located
[20][21][22][23][42,43,44,45]. This generates a great heterogeneity in PCs, and the context where they are located determines PC function, morphology, and surface markers
[24][25][46,47].
In the brain, PCs constitute the blood–brain barrier (BBB) along with endothelial cells (ECs) and are involved in various protective functions
[19][41]. Among their functions, PCs can participate in processes with stem cell-like properties, such as angiogenesis
[26][27][48,49]. They can also perform immune functions acting as macrophages and controlling T-cell migration by secreting different cytokines
[28][50]. PCs can phagocyte and act as antigen-presenting cells, overexpressing major histocompatibility complex (MHC) class I and II molecules, among others, to control T cell responses
[29][30][51,52].
Thus far, PCs might be considered as tumor attackers, but GB cells can manipulate PC functions, changing their phenotype to promote pro-tumoral instead of anti-tumoral immune responses
[10][31][32][33][17,39,53,54]. Thus, a detailed dissection and a better understanding of the role of CMA in PCs in a healthy brain versus those located in the tumor microenvironment is necessary to develop therapies to counter GB progression mediated by CMA-driven PCs. Tumor cell infiltration and invasion of the brain parenchyma, which provides a proliferative environment, can occur via interaction between tumor cells and parenchyma cells such as PCs
[10][17], creating a large network of microtubes and nanotubes that favors tumor development
[9][34][16,55]. In addition, different cancer stem cells (CSCs) are also found in the TME, making GB difficult to treat
[35][36][37][38][38,56,57,58]. Thus, the study of the TME, including the perivascular area with peritumoral PCs, is essential to make a good diagnosis of tumor progression.
2. Glioblastoma-Induced Chaperone-Mediated Autophagy in Pericyte
2. GB-Induced CMA in PC
In recent years, GB environment and implicated cells and molecules have acquired a special interest. In particular, peritumoral PCs are an attractive target to generate new therapeutical approaches because they are located in small blood vessels of the brain parenchyma, where GB spreads and invades the perivascular space
[10][31][17,39].
We have shown that GB interaction with PCs located in peritumoral areas is accompanied by a high expression of the CMA receptor LAMP-2A in both cells. In addition,
reseaour
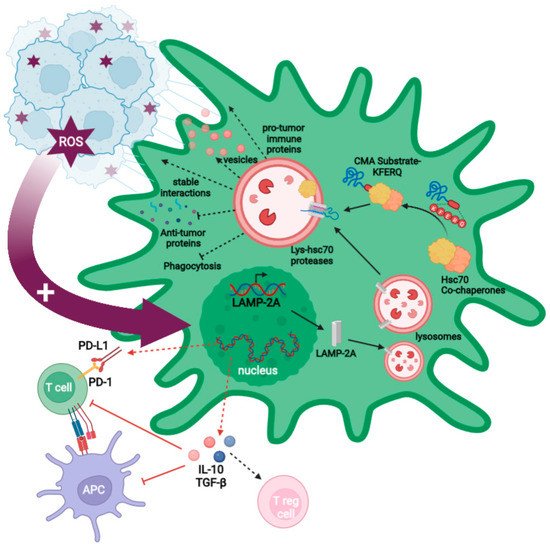
chers' data demonstrated that GB cells alter the PC proteome through aberrant degradation of critical proteins by CMA, promoting tumor survival and growth. A transient ROS burst is produced in the PCs following physical interaction with the GB cell, leading to overexpression of LAMP-2A, the only limiting molecule for CMA activity (
Figure 2). CMA is then aberrantly upregulated in PCs, consequently affecting the PC phenotype, which promotes stable cell-to-cell interactions that favor tumor cell proliferation, and which induces an immunosuppressive function that impairs anti-tumor immune responses
[10][31][17,39].
Figure 2. Model of the effects of GB-induced CMA on PCs. GB interaction with PCs promotes a ROS burst that triggers the increased upregulation of CMA in PCs through the expression of LAMP-2A (big garnet arrow). KFERQ motifs in CMA substrate proteins are recognized by the Hsc70 chaperone in a complex with other co-chaperones. This complex binds to substrates and carries them to the lysosomal membrane where they interact with LAMP-2A, and the lysosomal Hsc70 to translocate them into the lysosomal lumen to be degraded. Aberrant induction of CMA by GB leads to changes in the PC proteome, promoting an immunosuppressive function in PCs that exerts a negative regulation on T cells and antigen-presenting cells as a result of changes in gene expression programs that up-regulate the anti-inflammatory cytokines TGF-β and IL-10 (red dashed arrows). The expression of PD-L1 (negative regulator) and the lack of CD80 and CD86 (co-stimulatory molecules) promote regulatory T cell generation (red arrows). GB-induced CMA in PCs also facilitates tumor growth by stabilizing GB–PC interaction and preventing the secretion of anti-tumor proteins and phagocytic ability. In addition, it induces pro-tumor immune molecules and modulates the MSC-like properties such as the increase in the microvesicles secretion with pro-regenerative factors and cytokines that assist tumor proliferation (black dashed arrows).
Importantly, peritumoral PCs also seem to participate in the co-optation mechanism of GB, where the tumor does not create new blood vessels (angiogenesis) but kidnaps the existing ones to promote tumor cell infiltration and GB development
[39][40][62,63].
In addition, a recent RNA-seq study from
theour lab has shown that the tumor cell–PC interaction affects other functions in the PCs that might play important roles in GB progression. Supporting previous data,
theour study has revealed that GB-induced CMA in PCs promotes changes in gene expression pathways related to angiogenesis, cell adhesion, and immune and inflammatory functions of PCs. In fact, one of the most affected gene pathways in PCs interacting with GB was related to the phagosome, allowing
researchersus to identify the phagocytic function of PCs affected by CMA
[31][39].
In conclusion, GB cell–PC interaction leads to aberrant increased CMA activity in PCs (peritumoral and probably intratumoral), leading to major proteome changes in these cells. These promote the transformation of PCs from pro-inflammatory to anti-inflammatory cells and change other functions that affect the TME. As a consequence, the tumor escapes from anti-tumor immune responses and progresses (Figure 1). Thus, GB-induced CMA in PCs might be considered as a candidate therapeutical target to treat GB cancer.
2.1. GlioblastomaB-Induced Chaperone-Mediated Autophagy in PericyteMA in PC Is Required for GlioblastomaB Immune Evasion
One of the most important CMA-mediated changes in PCs following interaction with GB is the acquisition of an anti-inflammatory phenotype. GB-conditioned PCs increase the secretion of anti-inflammatory cytokines (e.g., IL-10 o TGF-β) (
Figure 2), while GB cells prevent an increased secretion of granulocyte–macrophage colony-stimulating factor (GM-CSF) related to their survival
[10][32][17,53].
GB-induced CMA is also responsible for the reduced expression of co-stimulatory molecules and MHC-II surface molecules in PCs, affecting antigen presentation and, therefore, impairing T cell activation, which, in turn, fails the anti-tumor T cells responses (
Figure 2)
[10][32][41][17,30,53]. Moreover, these changes have been recently corroborated by a transcriptomic study with CMA-deficient PCs, showing that inflammatory and immune gene expression pathways are affected by GB-induced CMA. These data revealed that PCs prevent GB elimination by their phagocytic capacity and the secretion of anti-tumor immune molecules, via GB-induced CMA
[31][39]. In addition, these data showed that GB-induced CMA in PCs also leads to the secretion of molecules that can promote pro-tumor immune responses, such as gelsolin, periostin, and osteopontin, supporting previous data showing the contribution of the PC secretome to GB immune evasion
[10][31][17,39].
2.2. GlioblastomaB-Induced Chaperone-Mediated AutophagyMA Upregulation in PericyteC Is Required for Stable Pericyte–GlioblastomaC–GB Interactions
GB–PC interactions lead to a functional network of connections that facilitate nutrient support and GB proliferation
[9][10][34][16,17,55]. GB-induced CMA upregulation is necessary to create the stable interactions between GB cells and PCs that maintain tumor cell survival and tumor growth. Those stable interactions are formed by nanotubes originated in PCs to GB cells and the regulation of adhesion protein expression (e.g., occludin), which is also dependent on GB-induced CMA
[10][31][17,39].
Using a GB mouse model with grafted CMA-deficient PCs,
rwe
searchers have previously shown a decrease in GB–PC interactions and impaired tumor formation when mice were infiltrated with GB cells
[10][17]. The characterization of cell-to-cell interactions in CMA-deficient PCs showed an absence of stable interactions and a subsequent reduction in GB cell survival. By contrast, mice grafted with control GB-conditioned PCs showed stable PC–GB interactions and tumor cell proliferation. Further electron microscopy analyses in PCs revealed a microvesicle secretion pattern dependent on GB-induced CMA. Interestingly, this vesicle secretion was mainly observed in the zone where the GB cell interacts with the PC
[10][17].
2.3. GlioblastomaB-Induced Chaperone-Mediated Autophagy in PericyteMA in PC Is Required to Modulate Pericyte PC Secretome and Pericyte Mesenchymal Stem Cell PC MSC-like Properties That Favor Tumor Progression
GB-induced CMA in PCs protects GB from a toxic PC secretome by inhibiting secretion of antitumor effectors (e.g., sparc, antithrombin, lumican, and vitamin D), which could be capable of destabilizing and inhibiting GB–PC interactions, causing GB cell death
[10][31][17,39]. By contrast, GB-induced CMA in PCs induces a secretome with high levels of proteins that can promote tumor angiogenesis and facilitate pro-tumor immune responses (e.g., angiotensin, gelsolin, periostin, and osteopontin)
[31][39].
Furthermore, GB-induced CMA in PCs affects their mesenchymal properties, facilitating tumor angiogenesis and progression. The expression of different angiogenic, angiotrophic, or anti-inflammatory factors changes when PCs interact with GB cells
[10][17]. Two of these factors are IL-6, a cytokine with regenerative properties, and the angiogenic factor VEGF, both highly secreted in GB-conditioned PCs
[32][53]. In addition, PC proliferation is reduced in response to GB-induced CMA, correlating with a loss of mitochondrial mass and the modulation of markers associated with mesenchymal properties (e.g., Sca-1, CD105, and CD90)
[10][17].