Clostridioides difficile is considered a nosocomial pathogen that flares up in patients exposed to antibiotic treatment. However, four out of ten patients diagnosed with C. difficile infection (CDI) acquired the infection from non-hospitalized individuals, many of whom have not been treated with antibiotics. Treatment of recurrent CDI (rCDI) with antibiotics, especially vancomycin (VAN) and metronidazole (MNZ), increases the risk of experiencing a relapse by as much as 70%. Fidaxomicin, on the other hand, proved more effective than VAN and MNZ by preventing the initial transcription of RNA toxin genes. Alternative forms of treatment include quorum quenching (QQ) that blocks toxin synthesis, binding of small anion molecules such as tolevamer to toxins, monoclonal antibodies, such as bezlotoxumab and actoxumab, bacteriophage therapy, probiotics, and fecal microbial transplants (FMTs).
- colonization
- toxin production
- antibiotics
- Clostridioides difficile
- biofilm formation
1. Introduction
2. Colonization of C. difficile to Human Intestinal Cells
CDI is contracted through the ingestion of endospores [18]. Germination of spores is controlled by the concentration and type of primary bile salts in the upper part of the GIT [19,20][19][20]. Chenodeoxycholate (CDCA) represses spore germination, whereas cholate (CA) induces germination [20]. Most of the primary bile acids (95%), conjugated with taurine and glycine or unconjugated, are absorbed in the terminal ileum and through the hepatic system [19,21][19][21]. Primary bile acids that reach the large intestine are converted by gut microbiota into secondary bile acids, for example, ω-muricholate (ωMCA), hyodeoxycholate (HDCA), ursodeoxycholate (UDCA), lithocholate (LCA), and deoxycholate (DCA) [19,22][19][22]. Physiological conditions, such as an excess of fermentable carbohydrates or an increase in deoxycholate (DOC, Figure 1), stimulate C. difficile to form biofilms in the human GIT, which may lead to recurrent episodes of CDI [23,24][23][24]. Biofilm formation is also regulated by quorum sensing (QS) signals such as cyclic diguanosine monophosphate (c-di-GMP; Figure 1).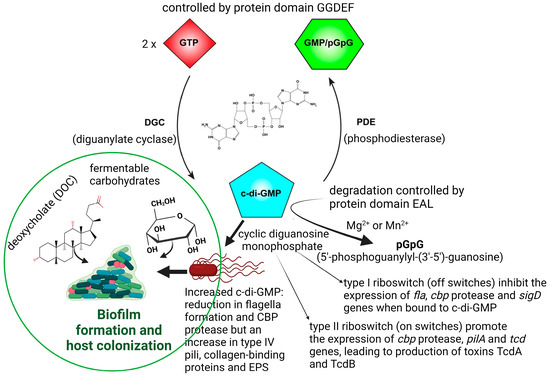
References
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeño-Tárraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786.
- Merrigan, M.; Venugopal, A.; Mallozzi, M.; Roxas, B.; Viswanathan, V.K.; Johnson, S.; Gerding, D.N.; Vedantam, G. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J. Bacteriol. 2010, 192, 4904–4911.
- Aliente, E.; Dawson, L.F.; Cairns, M.D.; Stabler, R.A.; Wren, B.W. Emergence of new PCR ribotypes from the hypervirulent Clostridium difficile 027 lineage. J. Med. Microbiol. 2012, 61, 49–56.
- Lessa, F.C.; Winston, L.G.; McDonald, L.C. Emerging Infections Program C. difficile Surveillance Team. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 2015, 372, 2369–2370.
- Chitnis, A.S.; Holzbauer, S.M.; Belflower, R.M.; Winston, L.G.; Bamberg, W.M.; Lyons, C.; Farley, M.M.; Dumyati, G.K.; Wilson, L.E.; Beldavs, Z.G.; et al. Epidemiology of community-associated Clostridium difficile infection, 2009 through 2011. JAMA Intern. Med. 2013, 173, 1359–1367.
- Gould, L.H.; Limbago, B. Clostridium difficile in food and domestic animals: A new foodborne pathogen. Clin. Infect. Dis. 2010, 51, 577–582.
- Hensgens, M.P.; Keessen, E.C.; Squire, M.M.; Riley, T.V.; Koene, M.G.; de Boer, E.; Lipman, L.J.; Kuijper, E.J. Clostridium difficile infection in the community: A zoonotic disease? Clin. Microbiol. Infect. 2012, 18, 635–645.
- Vedantam, G.; Clark, A.; Chu, M.; McQuade, R.; Mallozzi, M.; Viswanathan, V.K. Clostridium difficile infection: Toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes. 2012, 3, 121–134.
- Mada, P.K.; Alam, M.U. Clostridioides difficile Infection. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK431054/ (accessed on 23 January 2023).
- Okafor, C.M.; Clogher, P.; Olson, D.; Niccolai, L.; Hadler, J. Trends in and risk factors for recurrent Clostridioides difficile infection, New Haven County, Connecticut, USA, 2015–2020. Emerg. Infect. Dis. 2023, 29, 877–887.
- Miranda-Katz, M.; Parmar, D.; Dang, R.; Alabaster, A.; Greenhow, T.L. Epidemiology and risk factors for community associated Clostridioides difficile in children. J. Pediatr. 2020, 221, 99–106.
- Al-Jumaili, I.J.; Shibley, M.; Lishman, A.H.; Record, C.O. Incidence and origin of Clostridium difficile in neonates. J. Clin. Microbiol. 1984, 19, 77–78.
- Eglow, R.; Pothoulakis, C.; Itzkowitz, S.; Israel, E.J.; O’Keane, C.J.; Gong, D.; Gao, N.; Xu, Y.L.; Walker, W.A.; LaMont, J.T. Diminished Clostridium difficile toxin A sensitivity in newborn rabbit ileum is associated with decreased toxin A receptor. J. Clin. Investig. 1992, 90, 822–829.
- Bassis, C.M.; Theriot, C.M.; Young, V.B. Alteration of the murine gastrointestinal microbiota by tigecycline leads to increased susceptibility to Clostridium difficile infection. Antimicrob. Agents Chemother. 2014, 58, 2767–2774.
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. Host-Microbe Biol. 2016, 1, e00045-15.
- Mathur, H.; Rea, M.C.; Cotter, P.D.; Hill, C.; Ross, R.P. The efficacy of thuricin CD, tigecycline, vancomycin, teicoplanin, rifampicin and nitazoxanide, independently and in paired combinations against Clostridium difficile biofilms and planktonic cells. Gut Pathog. 2016, 8, 20.
- James, G.A.; Chesnel, L.; Boegli, L.; Pulcini, E.D.; Fisher, S.; Stewart, P.S. Analysis of Clostridium difficile biofilms: Imaging and antimicrobial treatment. J. Antimicrob. Chemother. 2017, 73, 102–108.
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Primers 2016, 2, 16020.
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259.
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512.
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174.
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap–Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498.
- Dawson, L.F.; Valiente, E.; Faulds-Pain, A.; Donahue, E.H.; Wren, B.W. Characterisation of Clostridium difficile biofilm formation, a role for Spo0A. PLoS ONE 2012, 7, e50527.
- Dubois, T.; Tremblay, Y.D.N.; Hamiot, A.; Martin-Verstraete, I.; Deschamps, J.; Monot, M.; Briandet, R.; Dupuy, B. A microbiota generated bile salt induces biofilm formation in Clostridium difficile. NPJ Biofilms Microbiomes 2019, 5, 14.
- McKee, R.W.; Mangalea, M.R.; Purcell, E.B.; Borchardt, E.K.; Tamayo, R. The second messenger cyclic Di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J. Bacteriol. 2013, 195, 5174–5185.
- Paul, R.; Weiser, S.; Amiot, N.C.; Chan, C.; Schirmer, T.; Giese, B.; Jenal, U. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 2004, 18, 715–727.
- Simm, R.; Morr, M.; Kader, A.; Nimtz, M.; Römling, U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 2004, 53, 1123–1134.
- Chan, C.; Paul, R.; Samoray, D.; Amiot, N.C.; Giese, B.; Jenal, U.; Schirmer, T. Structural basis of activity and allosteric control of diguanylate cyclase. Proc. Natl. Acad. Sci. USA 2004, 101, 17084–17089.
- Christen, M.; Christen, B.; Folcher, M.; Schauerte, A.; Jenal, U. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 2005, 280, 30829–30837.
- Aubry, A.; Hussack, G.; Chen, W.; KuoLee, R.; Twine, S.M.; Fulton, K.M.; Foote, S.; Carrillo, C.D.; Tanha, J.; Logan, S.M. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect. Immun. 2012, 80, 3521–3532.
- Lee, E.R.; Baker, J.L.; Weinberg, Z.; Sudarsan, N.; Breaker, R.R. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 2010, 329, 845–848.
- McKee, R.W.; Aleksanyan, N.; Garrett, E.M.; Tamayo, R. Type IV pili promote Clostridium difficile adherence and persistence in a mouse model infection. Infect. Immun. 2018, 86, e00943-17.
- Purcell, E.B.; McKee, R.W.; McBride, S.M.; Waters, C.M.; Tamayo, R. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J. Bacteriol. 2012, 194, 3307–3316.
- McKee, R.W.; Harvest, C.K.; Tamayo, R. Cyclic diguanylate regulates virulence factor genes via multiple riboswitches in Clostridium difficile. mSphere 2018, 3, e00423-18.
- Dingle, T.C.; Mulvey, G.L.; Armstrong, G.D. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect. Immun. 2011, 79, 4061–4067.
- Tulli, L.; Marchi, S.; Petracca, R.; Shaw, H.A.; Fairweather, N.F.; Scarselli, M.; Soriani, M.; Leuzzi, R. CbpA: A novel surface exposed adhesin of Clostridium difficile targeting human collagen. Cell Microbiol. 2013, 15, 1674–1687.
- Bordeleau, E.; Purcell, E.B.; Lafontaine, D.A.; Fortier, L.C.; Tamayo, R.; Burrus, V. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile. J. Bacteriol. 2015, 197, 819–832.
- Dawson, L.F.; Peltier, J.; Hall, C.L.; Harrison, M.A.; Derakhshan, M.; Shaw, H.A.; Fairweather, N.F.; Wren, B.W. Extracellular DNA, cell surface proteins and c-di-GMP promote biofilm formation in Clostridioides difficile. Sci. Rep. 2021, 5, 3244.
- Maldarelli, G.A.; Piepenbrink, K.H.; Scott, A.J.; Freiberg, J.A.; Song, Y.; Achermann, Y.; Ernst, R.K.; Shirtliff, M.E.; Sundberg, E.J.; Donnenberg, M.S.; et al. Type IV pili promote early biofilm formation by Clostridium difficile. Pathog. Dis. 2016, 74, ftw061.
- Stabler, R.A.; He, M.; Dawson, L.; Martin, M.; Valiente, E.; Corton, C.; Lawley, T.D.; Sebaihia, M.; Quail, M.A.; Rose, G.; et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009, 10, R102.
- Poquet, I.; Saujet, L.; Canette, A.; Monot, M.; Mihajlovic, J.; Ghigo, J.-M.; Soutourina, O.; Briandet, R.; Martin-Verstraete, I.; Dupuy, B. Clostridium difficile biofilm: Remodeling metabolism and cell surface to build a sparse and heterogeneously aggregated architecture. Front. Microbiol. 2018, 9, 2084.
- Taggart, M.G.; Snelling, W.J.; Naughton, P.J.; La Ragione, R.M.; Dooley, J.S.G.; Ternan, N.G. Biofilm regulation in Clostridioides difficile: Novel systems linked to hypervirulence. PLoS Pathog. 2021, 17, e1009817.
- Valiente, E.; Bouché, L.; Hitchen, P.; Faulds-Pain, A.; Songane, M.; Dawson, L.F.; Donahue, E.; Stabler, R.A.; Panico, M.; Morris, H.R.; et al. Role of glycosyltransferases modifying type B flagellin of emerging hypervirulent Clostridium difficile lineages and their impact on motility and biofilm formation. J. Biol. Chem. 2016, 291, 25450–25461.
- Ðapa, T.; Leuzzi, R.; Ng, Y.K.; Baban, S.T.; Adamo, R.; Kuehne, S.A.; Scarselli, M.; Minton, N.P.; Serruto, D.; Unnikrishnan, M. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 2013, 195, 545–555.
- Purcell, E.B.; McKee, R.W.; Bordeleau, E.; Burrus, V.; Tamayo, R. Regulation of type IV pili contributes to surface behaviors of historical and epidemic strains of Clostridium difficile. J. Bacteriol. 2016, 198, 565–577.
- Pedrido, M.E.; de Oña, P.; Ramirez, W.; Leñini, C.; Goñi, A.; Grau, R. Spo0A links de novo fatty acid synthesis to sporulation and biofilm development in Bacillus subtilis. Mol. Microbiol. 2013, 87, 348–367.
- Walter, B.M.; Cartman, S.T.; Minton, N.P.; Butala, M.; Rupnik, M. The SOS response master regulator LexA is associated with sporulation, motility and biofilm formation in Clostridium difficile. PLoS ONE 2015, 10, e0144763.
- Chu, M.; Mallozzi, M.J.; Roxas, B.P.; Bertolo, L.; Monteiro, M.A.; Agellon, A.; Viswanathan, V.K.; Vedantam, G. A Clostridium difficile cell wall glycopolymer locus influences bacterial shape, polysaccharide production and virulence. PLoS Pathog. 2016, 12, e1005946.
- Jain, S.; Smyth, D.; O’Hagan, B.M.G.; Heap, J.T.; McMullan, G.; Minton, N.P.; Ternan, N.G. Inactivation of the dnaK gene in Clostridium difficile 630 Δerm yields a temperature-sensitive phenotype and increases biofilm forming ability. Sci. Rep. 2017, 7, 17522.
- Pantaléon, V.; Soavelomandroso, A.P.; Bouttier, S.; Briandet, R.; Roxas, B.; Chu, M.; Collignon, A.; Janoir, C.; Vedantam, G.; Candela, T. The Clostridium difficile protease Cwp84 modulates both biofilm formation and cell-surface properties. PLoS ONE 2015, 10, e0124971.
- Cuenot, E.; Garcia-Garcia, T.; Douche, T.; Gorgette, O.; Courtin, P.; Denis-Quanquin, S.; Hoys, S.; Tremblay, Y.D.N.; Matondo, M.; Chapot-Chartier, M.-P.; et al. The Ser/Thr kinase PrkC participates in cell wall homeostasis and antimicrobial resistance in Clostridium difficile. Infect. Immun. 2019, 87, e00005-19.
- Jain, S.; Graham, C.; Graham, R.L.J.; McMullan, G.; Ternan, N.G. Quantitative proteomic analysis of the heat stress response in Clostridium difficile strain 630. J. Proteome Res. 2011, 10, 3880–3890.
- Johnston, J.L.; Sloan, J.; Fyfe, J.A.M.; Davies, J.K.; Rood, J.I. The recA gene from Clostridium perfringens is induced by methyl methanesulphonate and contains an upstream Cheo box. Microbiology 1997, 143, 885–890.
- Dapa, T.; Unnikrishnan, M. Biofilm formation by Clostridium difficile. Gut Microbes. 2013, 4, 397–402.
- Slater, R.T.; Frost, L.R.; Jossi, S.E.; Millard, A.D.; Unnikrishnan, M. Clostridioides difficile LuxS mediates inter-bacterial interactions within biofilms. Sci. Rep. 2019, 9, 9903.
- Berges, M.; Michel, A.-M.; Lassek, C.; Nuss, A.M.; Beckstette, M.; Dersch, P.; Riedel, K.; Sievers, S.; Becher, D.; Otto, A.; et al. Iron Regulation in Clostridioides difficile. Front. Microbiol. 2018, 9, 3183.
- Deshpande, A.; Olaitan, A.O.; Mckelvey, A.M.; Rutherford, T.J.T.; Hurdle, J.G. The ferrous iron transporter FeoB1 is essential for Clostridioides difficile toxin production and Pathogenesis in Mice. bioRxiv. 2022.
- Ho, T.D.; Ellermeier, C.D. Ferric uptake regulator for control of putative iron acquisition systems in Clostridium difficile. J. Bacteriol. 2015, 197, 2930–2940.
- Yamaki, J.; Chawla, S.; Tong, S.; Lozada, K.A.; Yang, S. Iron effects on Clostridioides difficile toxin production and antimicrobial susceptibilities. Antibiotics 2022, 11, 537.
- Semenyuk, E.G.; Laning, M.L.; Foley, J.; Johnston, P.F.; Knight, K.L.; Gerding, D.N.; Driks, A. Spore formation and toxin production in Clostridium difficile biofilms. PLoS ONE 2014, 9, e87757.
- Nale, J.Y.; Shan, J.; Hickenbotham, P.T.; Fawley, W.N.; Wilcox, M.H.; Clokie, M.R. Diverse temperate bacteriophage carriage in Clostridium difficile 027 strains. PLoS ONE 2012, 7, e37263.
- Ibanez de Aldecoa, A.L.; Zafra, O.; Gonzalez-Pastor, J.E. Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front. Microbiol. 2017, 8, 1390.
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The quorum sensing inhibitor hamamelitannin increases antibiotic susceptibility of Staphylococcus aureus biofilms by affecting peptidoglycan biosynthesis and eDNA release. Sci. Rep. 2016, 6, 20321.
- Svensson, S.L.; Pryjma, M.; Gaynor, E.C. Flagella-mediated adhesion and extracellular DNA release contribute to biofilm formation and stress tolerance of Campylobacter jejuni. PLoS ONE 2014, 9, e106063.
- Sekulovic, O.; Fortier, L.C. Global transcriptional response of Clostridium difficile carrying the CD38 prophage. Appl. Environ. Microbiol. 2015, 81, 1364–1374.
- Yoon, S.; Yu, J.; McDowell, A.; Kim, S.H.; You, H.J.; Ko, G. Bile salt hydrolase-mediated inhibitory effect of Bacteroides ovatus on growth of Clostridium difficile. J. Microbiol. 2017, 55, 892–899.
- Macy, J.M.; Ljungdahl, L.G.; Gottschalk, G. Pathway of succinate and propionate formation in Bacteroides fragilis. J. Bacteriol. 1978, 134, 84–91.