Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Ulrich Gergs.
Glucagon exerts effects on the mammalian heart. These effects include alterations in the force of contraction, beating rate, and changes in the cardiac conduction system axis. The cardiac effects of glucagon vary according to species, region, age, and concomitant disease. Depending on the species and region studied, the contractile effects of glucagon can be robust, modest, or even absent. Glucagon is detected in the mammalian heart and might act with an autocrine or paracrine effect on the cardiac glucagon receptors. The glucagon levels in the blood and glucagon receptor levels in the heart can change with disease or simultaneous drug application.
- glucagon
- glucagon receptor
- human heart
- mouse heart
1. Introduction
Glucagon is mainly formed in α-cells in the islets of the Langerhans within the human pancreas [1]. In the human liver, but to a lesser extent in the heart, glucagon augments gluconeogenesis and glycogenolysis. Therefore, glucagon acts as a functional antagonist of insulin: insulin decreases the blood concentrations of glucose, and glucagon increases the blood glucose levels.
Glucagon was one of the first stimuli identified as an adenosine-3′,5′-cyclic monophosphate (cAMP, Figure 1)-increasing agent (liver: [2]). The positive inotropic effects of glucagon in the heart were initially thought to result from the release of stored noradrenaline and subsequent stimulation of the β-adrenoceptors in the heart [3]. However, these positive inotropic effects and cAMP-increasing effects of glucagon in the heart were later shown not to be blocked by the β-adrenoceptor antagonist propranolol (dog: [4], cat, and human: [5]), and thus it is regarded as being mediated by a receptor of its own: the glucagon receptor. Yet, glucagon can also activate another receptor called the glucagon-like protein-1-receptor (GLP1-R, Figure 1). Indeed, glucagon-like peptide-1-receptors and related so-called glucagon-like protein-2-receptor are also expressed and functional in the heart [6,7][6][7]. Glucagon is not an agonist to the glucagon-like protein-2 receptor [8]. Accordingly, the glucagon-like protein-2-receptor will not be discussed herein.
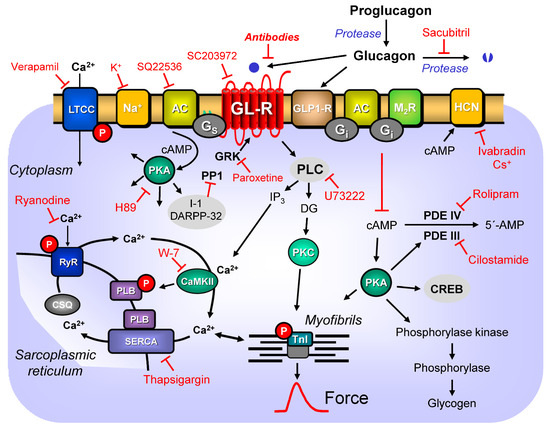
Figure 1. Potential mechanism(s) of action of glucagon in the human and mouse cardiomyocytes. Stimulation of the activity of the glucagon receptor (GL-R, blocked by antibodies or SC203972) by endogenous glucagon leads via stimulatory GTP-binding proteins (Gs) to an increase of adenylyl cyclase (AC) activity (inhibitable by SQ22536). AC increases the formation of 3′,5′-cyclic adenosine mono phosphate (cAMP) that stimulates cAMP-protein kinases (PKAs, inhibitable by H89). PKAs phosphorylate (red P) and thus activate phospholamban (PLB) at the amino acid serine 16, the inhibitory subunit of troponin (TnI), the ryanodine receptor (RYR), the L-type calcium channel (LTCC, inhibitable by verapamil), cAMP response element-binding proteins (CREB), or phosphorylase kinase (which then phosphorylates and activates phosphorylase to cleave glycogen). PKA also phosphorylates the so-called phosphatase inhibitors such as inhibitor 1 (I1) or dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP32) that then inhibit the activity of serine/threonine protein phosphatase 1 (PP1). PLB is also phosphorylated by calcium-calmodulin-dependent protein kinases (CaMKII: inhibited by W-7) on amino acid threonine 17. cAMP can directly activate the hyperpolarization-activated cyclic nucleotide-gated channels (HCN) in the sinus node (inhibited by ivabradin or cesium ions (Cs+)). The formed cAMP can be degraded to inactive 5′-adenosine mono phosphate (5′-AMP) by phosphodiesterases (PDE III: inhibited by cilostamide, PDE IV: inhibited by rolipram). Calcium cations (Ca2+) are stored on calsequestrin (CSQ) in the sarcoplasmic reticulum (SR) and are released via RYR (inhibitable by ryanodine) from the SR. The released Ca2+ bind to thin myofilaments and as a result systolic force is augmented. In cardiac diastole, concentrations of Ca2+ fall because Ca2+ are pumped into the SR via the SR-calcium ATPase (SERCA, inhibitable by thapsigargin). GL-R can be inactivated by G-protein-dependent protein kinases (GKR: inhibited by paroxetine). GL-R can activate phospholipase C (inhibited by U73222). Cardiac sodium channels (Na+) in the sarcolemma can be functionally inhibited by potassium ions (K+) that lead to partial depolarization. Proglucagon can be cleaved to glucagon. Glucagon is degraded by another protease (inhibited by sacubitril). Glucagon can also activate glucagon-like-protein-1-receptors (GLP1-R). Acetylcholine can activate M2-muscarinic receptors and thereby, via GTP-binding inhibitory proteins (Gi), reduce the activity of AC: see text for further details.
2. Glucagon Receptor
Initially, the rat glucagon receptor was cloned in the year 1993, thirty years ago [9]. The human glucagon receptor was cloned in 1994 and displayed an 82% protein sequence identity with the rat glucagon receptor [10]. The binding affinity of glucagon to the human glucagon receptor was reported to be 5 nano mole per liter (nM) of glucagon [10]. A wealth of data has defined the three-dimensional structure of the glucagon receptor and how the agonist glucagon or its analogs bind to this receptor [11,12,13,14][11][12][13][14]. This issue has been discussed elsewhere and is not the focus of the current review [15].
That the glucagon receptor is present in the mammalian heart, specifically in the rat heart, was already reported in the first cloning papers [9,16][9][16]. Using an RNase protection assay, Hansen et al. [17] quantified the messenger ribonucleotides (mRNA) for glucagon in a rat heart. In their hands, the mRNA expression of the glucagon receptor in the heart was about 50% of the mRNA expression of the glucagon receptor in the rat liver [17]. This relatively strong expression may indicate an essential function of glucagon receptors in the heart. Later, the glucagon receptor was found in mRNA levels in the mouse heart [18]. The glucagon receptor as mRNA was highly expressed in the mouse’s right atrium, but very lowly expressed in the left atrium or cardiac ventricle [19]. One would predict that the contractile function of the glucagon receptor would follow its regional expression, which is, to some extent, apparently the case (see below for the rat heart). On a protein level, using radioactive glucagon and autoradiography, glucagon receptors have also been detected in the mouse heart, suggesting a possible functional role of the glucagon receptor also in the mouse heart [20].
The human glucagon receptor comprises only 477 amino acids, whereas the mouse and rat glucagon receptors comprise 485 amino acids [16,21][16][21]. In human glucagon-receptor-transfected cells, glucagon stimulates adenylyl cyclase (AC) very potently, with a half maximally stimulatory concentration (EC50-values) of 10 pico mole per liter (10 pM) [21]. This high potency of glucagon in the stimulation of the glucagon receptor is important to keep in mind, because this potency is much higher than the concentration of glucagon required to raise the beating rate or force of contraction in the mammalian heart.
The human glucagon receptor is located on chromosome 17 at 17q25 [21]. It may be relevant that the protein sequence of the human glucagon receptor is not only shorter than that of the mouse glucagon receptor (see above), but there is only about an 80% protein sequence identity between the human and mouse glucagon receptors [22]. This may translate into species differences in glucagon receptor function. Therefore, arguments can be made that the normal mouse heart might not be the best model for understanding the human cardiac glucagon receptor. This concern for the species differences of the glucagon receptor has prompted the development of transgenic mouse models, where the human receptor is expressed instead of the endogenous mouse receptor in the mouse heart and functional differences are searched for (see below).
The mRNA of the glucagon receptor has been detected in all regions of the human heart [7]. However, the expression of the human glucagon receptor at the mRNA level is very heterogeneous with respect to the region of the heart, but also with respect to the patient being studied. For instance, in one study, no expression of glucagon receptors was found using reverse transcriptase polymerase chain reaction (RT-PCR) in the left human ventricle, and in only 2 of 15 different patient samples was the expression of the glucagon receptor in the right ventricle noted with RT-PCR, while in 3 of 15 patient samples with RT-PCR, the expression of the glucagon receptor was noted in the right atrium. The glucagon receptor was detectable in 1 of 15 patient samples in the left atrium [7]. Other researchers, using different patients and slightly different methodologies, failed to detect any expression at the mRNA level or protein level (Western blotting) of the glucagon receptor in samples from the heart (left atrium, right atrium, left ventricle, right ventricle, and sinus node, [23]): they studied samples from a total of ten patients; this included diseased patients (e.g., hypertrophic obstructive cardiomyopathy or autoimmune myocarditis), but also tissue from donors that had died from accidents (remarkably, only two donors did not take any drugs, which might have affected the glucagon receptor expression in the heart). The donor tissues were in asystole for a mean period of 83 min. This might have led to degradation of the mRNA for the glucagon receptor [23]. Likewise, others have failed to detect mRNA of the glucagon receptor in the human heart [24]. They studied cardiac tissue from over 100 donors [24]. They used atrial tissue (left or right atrium was not specified) and left ventricular tissue from patients [24]. Moreover, these cardiac samples were studied post mortem [24]. Hence, one could argue that the mRNA for the glucagon receptor from the cardiac tissue may have been degraded during processing and, therefore, the mRNAs for the glucagon receptor in the hearts were not detected [23,24][23][24]. This assumption is not very far fetched: the glucagon receptor has been functionally found in human coronary arteries (Table 1). Hence, when mRNA is prepared from the whole human heart, at least these vascular glucagon receptors should have been detectable in the mRNA prepared from the whole human heart samples. As this was not the case in these studies [23[23][24],24], the possible degradation of the mRNA of the glucagon receptor cannot be completely ruled out as a limitation of these studies [23,24][23][24].
Table 1.
Species- and age-dependency of glucagon-induced increases in cardiac contractility in mammals, including humans.
| Left Atrium | Right Atrium | Ventricle | Remarks | |
|---|---|---|---|---|
| Cat | [3,5,25][3][5][25] PCE [25] PIE |
[5,25,26][5][25][26] In vivo: PIE [5,27][5][27] PIE: papillary muscle [27] PIE: perfused heart [28] No PIE in heart failure [25,29][25][29] PIE in heart failure |
||
| Dog | [3,4][3][4] PIE | [3,4,30,31,32,33,34,35,][3]36,[4]37,[38,39,30][31][32][33 |
The glucagon-stimulated generation of cAMP has similar EC50-values (half maximum concentrations of stimulation) in mouse livers as those in humanized mouse livers (40 nM glucagon and 13 nM glucagon, respectively, [91]). In contrast, in human liver membranes, glucagon-stimulated cAMP formation has an affinity of 6 nM [92]. These values are somewhat higher than the physiological levels of glucagon in the blood. However, from these results, one would predict similar EC50 values (that is, 13 nM to 40 nM) for the positive inotropic and positive chronotropic effects of the glucagon in muscle strips from experimental animals or human muscle strips if the human cardiac glucagon receptor mediates the contractile effects of the glucagon in the human heart. However, this is not the case under all conditions, and this issue will be addressed in more depth below.
More recently, systematic efforts have been made to identify the glucagon receptor with a battery of antibodies. Using tissue from glucagon receptor knockout (KO) mice as a negative control, two out of twelve commercially available receptor antibodies were identified that could detect the glucagon receptor in Western blots at about 55 kDa [24]. Only one of the twelve antibodies was specific in fixed liver sections [24]. With this specific antibody in their hands, researchers could identify the glucagon receptor in the immunohistochemistry of the mouse heart on cardiomyocytes [24]. Apparently, they did not study with this specific antibody the expression of the glucagon receptors in the human heart [24], which would have been interesting given the discrepancies in the mRNA expression data for the glucagon receptors in the human heart, as discussed above.
There is speculation in the literature that not all cardiac effects of glucagon can be explained by its cognate glucagon receptor or by its cross-reactivity to the glucagon-like peptide 1 receptor. Still, there might be an unknown orphan glucagon receptor in the human genome [93]. Hence, this chapter might not yet be closed.
Autoradiography with radioactively labeled glucagon has failed to detect a specific signal in the human heart [24]. This has been discussed as to possibly indicate that the glucagon receptor is lacking in the human heart, because radioactive glucagon is expected to bind to the glucagon receptor and thus induce a signal in autoradiography, indicative of the presence of the glucagon receptor [24]. This lack of signal in autoradiography might mean that the glucagon receptors are not present on the surface of cardiomyocytes, but are mainly in the cytosol, where they cannot react with radioactively labeled glucagon, but could react with an antibody [24].
As mentioned above, glucagon has about 140 times less affinity for glucagon-like peptide receptors (about 130 nM) than the specific agonists of these receptors. GLP-1 receptor expression as mRNA was three times more abundant in the human atrium than in the ventricle or cardiomyocytes from the human left ventricle [94]. In isolated, electrically driven atrial muscle strip preparations from ten patients, a GLP-1 receptor agonist (6 nM and higher of exenatide, a selective GLP-1 receptor agonist that does not activate the glucagon receptors, Table 3) concentration- and time-dependently raised the force of contraction [94]. It is interesting to note, but not readily understood, that only in 2 of 14 ventricular muscle strips from non-failing human (donor) hearts, did exenatide exert a positive inotropic effect. In contrast, all the human atrial and human ventricular samples in this study expressed the mRNA of the GLP-1 receptors [94]. GLP-1 receptor expression as a protein in the human heart was not reported [94]. Therefore, perhaps in the ventricular tissue, the protein expression of GLP-1 is usually less in the human ventricle than that in the human atrium, which may explain this discrepancy in function [94]. If we assume the lowest estimate of the EC50-value of 6 nM for the GLP-1 receptor, and given that glucagon is 100 times less potent at the GLP-1 receptor than exenatide is, the lowest EC50 estimate of glucagon at the GLP-1 receptor would be 600 nM. Therefore, a positive inotropic effect occurring at around 600 nM to 1 μM of glucagon (reported in many studies on cardiac effects on the force of contraction in human hearts, shown in Table 1) could mean that these effects are mediated by glucagon acting through the GLP-1 receptors. This should be studied directly by repeating such experiments. One could study in the same muscle strips exenatide and glucagon in head-to-head comparison. One could also include GLP1-receptor antagonists and, in comparison, also antagonists selective for glucagon receptors in such contraction experiments (possible antagonists have been selected in Table 3).
Table 3.
List of agonist, antagonists, and inhibitory antibodies available for the study of glucagon receptors in vitro and in vivo.
| Compound: | Affinity at GR |
Organism Cells |
Half Life | |
|---|---|---|---|---|
| Antagonist HM15136 |
[95] Human GR: EC50 = 92 pM | CHO, Mice | In mice: 136 h | |
| Antagonist: nonpeptide:2-(4-pyridyl)-5-(4-chlorophenyl)-3-(5-bromo-2-propyloxy-phenyl)pyrrole (L-168,049) | Human GR [96] IC50 = 3.7 nM | ][34][35][36][37][38][39]40[40] PCE [31,36][31][36] PIE |
[3,4,34,35,39,40][3][4][34][35][39][40] PIE [4] failing dog ventricle |
[40] Coronary perfusion enhanced |
| Embryon-ic chick heart | [41 | |||
| ] | ||||
| [ | ||||
| 54 | ||||
| ] | ||||
| [ | ||||
| 72 | ||||
| ] | ||||
| No PCE | ||||
PCE: positive chronotropic effect, PIE: positive inotropic effect, APD: action potential duration, and i.v.: intravenously, superscripts in table refer to references.
If these results [7,23,24][7][23][24] are representative and typical of the human heart and not just chance findings, the published contractile data are hard to reconcile with these mRNA expression data of the human cardiac glucagon receptor: in most samples from failing and non-failing ventricular muscle strips, glucagon exerted positive inotropic effects (Table 1). In the study by Baiio et al. [7], no contractile function of glucagon was measured in samples that were analyzed for the mRNA expression of the glucagon receptor in human heart tissue [7]. One would have predicted in their tissues [7] no positive inotropic effects of the glucagon in the atrial and ventricular muscle strips in which the mRNA of the glucagon receptor was measured, and no inotropic effects of glucagon in the samples where the mRNA for the glucagon receptor was below the detection limit.
Here, one must remember that the glucagon given to patients or animals might not only stimulate the glucagon receptor, but also the related GLP-1 receptor. While GLP-1 binds to the human GLP-1 receptor with an affinity of around 5 nM, glucagon is about 100 times less potent at binding to the human GLP-1 receptor [73].
In many studies (Table 1), 1–10 micro mole per liter (μM) of glucagon was given in an organ bath to stimulate the force of contraction in human cardiac muscle preparations. A total of 1 µM of glucagon should easily stimulate both the glucagon and GLP-1 receptors in the human heart (Table 2).
Table 2. Species- and age-dependency of signal transduction pathways used by glucagon in cardiac preparations from mammals, including humans.
| Species | Right Atrium | Left Atrium | Ventricle | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cat | [28] AC stimulation in normal heart [28] No stimulation of AC in failing hearts |
[74] AC stimulation [74] PDE not inhibited |
|||||||||||||||||||||||||||||
| Chicken embryonic ventricular cardio-myocytes | [75] Calcium transients increased | Mice | |||||||||||||||||||||||||||||
| ] | PCE | ||||||||||||||||||||||||||||||
| Dog | [31] | ||||||||||||||||||||||||||||||
| AC not stimulated | |||||||||||||||||||||||||||||||
| Antagonist: nonpeptide: N-[3-cyano-6-(1,1-dimethylpropyl)-4,5,6,7-tetrahydro-1-benzothien-2-yl]-2-ethylbutanamide (SC203972) | [ | [96] Human GR IC50 = 181 nM31] AC not stimulated |
[76] cAMP increase |
Mice | Frog | Embryonic chick heart | [42] PIE | ||||||||||||||||||||||||
| [ | 31 | ] | AC stimulated | [41] AC stimulated | [41] Glucagon binding increased with age | ||||||||||||||||||||||||||
| Antagonist, peptide: desHis1-Pro4-glucagon |
[97] Human GR IC50 = 1 nM |
Mice | persistent biological effects |
Guinea pig | [3,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57][3][43][ | Frog heart44 | |||||||||||||||||||||||||
| Antagonist, peptide, des-His1-[Glu9]glucagon, GR specific, | [77]][45][46][47][48][49][50][ PDE inhibition | [98]51][52][53][54][55][56][57] PIE | Rat GR[43,[44,45,7747,58,59][43]][44][45][47][58][59] PCE | LTCC stimulation[ | |||||||||||||||||||||||||||
| HEK | 44 | , | 45][44][45] No inotropic effect | ||||||||||||||||||||||||||||
| Human cardiac tissue (isolated) | [50 | Guinea pig ventricle] No inotropic effect | [45 | ||||||||||||||||||||||||||||
| LY2490921 GR unspecific | ,77][45][77] cAMP not increased | [23,50 | [59 | [98] Rat GR: 1.3 µM, [98] Human GLP-1-R: 1.2 µM][23][,76][59]50] No inotropic effect |
[76] AC not stimulated[50,60,61][50][60][61] No inotropic effect, [60] APD shortened, in vivo: PIE [49,62][49 |
[42][62] PIE in left ventricular papillary muscle strips from failing hearts | |||||||||||||||||||||||||
| , | 77 | HEK | ][42][77] PDE inhibition | Human patient or healthy volunteer In vivo |
[38,51,52,53,63,64][38][51][52][53][63][64] PCE | [38,3851,][5152,][5253,]63,64][[ | |||||||||||||||||||||||||
| Human atrium | 53 | ] | [ | ||||||||||||||||||||||||||||
| Antagonist, peptide desHis1Glu9(Lys30PAL)-glucagon |
[99] GR: 170 pM | 78] AC stimulation and inhibition | [63][64] PIE (1–5 mg i.v.) | [38,52,63,64][38][52][63][64] Vascular peripheral resistance decreased, [38,51,52,63][38][51][52][63] nausea [51,63][51][63] vomiting, [64] flushing, [64] palpitations, [64] diarrhoea, and [64] hyperglycemia [52] Coronary flow increased, [52] oxygen consumption increased, [52 |
] Blood glucose increased | ||||||||||||||||||||||||||
| Mice, HEK293 | persistent biological effects | ||||||||||||||||||||||||||||||
| Monkey | Human | ||||||||||||||||||||||||||||||
| Antibody REMD2.29 | [20] | [ | 42] PIE | ||||||||||||||||||||||||||||
| AC stimulation in normal adult heart, | [74] No stimulation of AC in adult failing hearts, [74] AC stimulation human fetal heart |
[ | [100] 30 pM79] AC stimulation, [79] PDE not inhibited |
Mouse adult |
|||||||||||||||||||||||||||
| Rabbit | [76] AC not stimulated in membranes[18] PCE, [54] No PCE |
[50 | [76] AC not stimulated in membranes, [80] AC stimulated in nuclei |
||||||||||||||||||||||||||||
| Glucagon: physiological agonist | [95] Human GR: 800 pM | ] | No PIE | ||||||||||||||||||||||||||||
| [ | 98] Rat GR: 400 pM [98] Rat GLP-1R: 4.9 nM |
[95] 5 min | Mouse fetal and neo-natal | Monkey | [81] PDE inhibition [42[65] PCE late-term fetal mouse heart |
]Neonatal mouse cardiomyocyte [54,65][54][65] PIE, [55 | |||||||||||||||||||||||||
| Sacubitril: inhibitor of glucagon degradation | AC not stimulated | [101] Inhibition of glucagon metabolism: about 1 nM [42] AC not stimulated |
sacubitrilat,[56,57][ | 42]55][56][57] PCE | |||||||||||||||||||||||||||
| AC not stimulated | Rabbit | [66] No PCE | [3,66][3][66] No effect | ||||||||||||||||||||||||||||
| Mouse | |||||||||||||||||||||||||||||||
| Anti-sense for GR | [77] PDE inhibition | ||||||||||||||||||||||||||||||
| [ | 102] Mice, [103] patients T2DM |
Rat, adult | [3] No inotropic effect [67, |
Neonatal rat heart68] | [72][67][68] PIE | [23,44,67,68,69,70][23][44][67][68][69][70] PCE [3] No inotropic effect |
No AC stimulation[3,44,[45,44][68,45][70,71][3]68][70][71 | ] | |||||||||||||||||||||||
| PIE | |||||||||||||||||||||||||||||||
| Antagonist: Exenadin 9–39 | [98] IC50: Human GLP-1-R: 17 nM | [ | 44 | ,68][44][68 | ] Relaxation shortened | ||||||||||||||||||||||||||
| Rat, fetal | Rat heart | [42,45,82,83][42][45][54, | |||||||||||||||||||||||||||||
| Exenadin4: GLP-1-receptor agonist | [98[82][83] AC stimulation | ] EC50: Human GLP-1-R: 30 pM[45,[457284]][84] LTCC stimulation
AC: activity of cardiac adenylyl cyclase, PDE: phosphodiesterase, cAMP: 3′,5′ cyclic adenosine monophosphate, and LTCC: L-type calcium ion channel.
GR: glucagon receptor, GLP-1: glucagon-like-protein-1, min: minutes, h = hours, EC50 = half maximally stimulatory concentration, IC50 = half maximally inhibitory concentration. pM: pico (10−12) mole per liter, nM: nano (10−9) mole per liter, and µM: micro (10−6) mole per liter. HEK: human embryonic kidney 293 cells, CHO: Chinese hamster ovary cells, and T2DM: type 2 diabetes mellitus.
However, the published data would be consistent with the assumption that, at least in the non-failing human heart, the positive inotropic effect of glucagon is mediated, in all likelihood, by two receptors. At low glucagon concentrations, glucagon would stimulate the glucagon receptors in human atrial and ventricular preparations. At higher glucagon concentrations, glucagon would stimulate the GLP-1 receptors. When glucagon stimulates the glucagon receptor, as well as when glucagon stimulates the GLP-1 receptor, an increase in the cAMP concentrations in cardiomyocytes would follow, and this would increase the force of contraction in any case (Figure 1).
Interestingly, as heart failure (detected clinically, patients aged from 18 years to 62 years) increased, the positive inotropic effect was weaker and was even absent in papillary muscle strips from patients with end-stage heart failure, and, in their samples, glucagon failed to increase the activity of AC [60].
As animal models, to better understand the role of the glucagon receptors, mice with a generalized knockout of the glucagon receptor or heart-specific knockdown of the glucagon receptor have been generated [18]; they have a cardiovascular phenotype [18] that will be discussed below. However, these mice would also be helpful in the future to study the inotropic and chronotropic effects of glucagon. One would predict that glucagon should be unable to increase the force of contraction in the atrium and/or ventricle of mice with a knockout of the glucagon receptor.
3. Glucagon Receptor RegulationIn principle, the expression of the glucagon receptor can be regulated, and thereby the function of glucagon can be changed in the heart. In isolated, cultivated adult rat hepatocytes after 24 h, the glucagon receptor (at the mRNA and protein level) is down-regulated in the presence of glucose and upregulated in the presence of cAMP-increasing agents, acting via the stimulation of adenylyl cyclase (AC) through forskolin, the activation of the glucagon receptor through glucagon, or the inhibition of cAMP degradation (using a phosphodiesterase (PDE) inhibitor, 3–isobutyl-1-methyl-xanthine) and the stimulation of the β-adrenoceptor (by isoprenaline, [104]). If we assume that the glucagon receptor stimulates AC, then the measurement of AC is a surrogate parameter of glucagon-receptor–protein expression. Under this limitation, one can try to interpret older data generated before cloning the glucagon receptor was described.
Experimental hypothyroidism reduces the efficacy (but not the potency) of glucagon to stimulate AC (Table 2) in the hearts of rats, conceivably via a reduction in the density of the glucagon receptors [105]. Chronic β-adrenergic stimulation with a parenteral injection of living rats with isoprenaline likewise reduces the efficacy (but not potency) of glucagon to increase the activity of cardiac AC [106]. In the hearts of obese or hypertensive rats, the efficacy of glucagon to stimulate the activity of AC is reduced. In the dog heart, glucagon is more effective in stimulating the activity of AC in the ventricle than the atrium [81]. In the monkey atrium, glucagon fails to activate AC [81]. In human atrial or ventricular preparations, 10 μM of glucagon increases the activity of AC [107]. The problem is that very high concentrations of glucagon are required to detect an increase in AC activity.
Nevertheless, artifacts should be ruled out. Hence, there is a clear need to also measure in protein levels the expressional changes alluded to above for the glucagon receptor, for example, in hypothyroidism or after prolonged β-adrenergic stimulation, to confirm or refute this earlier work.
Like other heptahelical receptors, the glucagon receptor exhibits homologous desensitization due to receptor downregulation. At least this could be concluded when rats were treated with injections of 0.5 milli gram (mg) of glucagon/150–200 g (g, body weight) over 10 days [108]. The density of the signal (the binding of the membranes from the rat liver to the radioactive glucagon) declined [108]. However, PCR or better Western blot data in the heart would also be interesting under these conditions to confirm this reduced glucagon receptor expression over time. In a guinea pig left atrium, 1 μM of glucagon increased the force and cAMP levels, and both these parameters were desensitized by a 15 min pre-treatment with 2 μM of glucagon [43]. Similarly, the desensitization of glucagon in cardiac contractility is well known [3,44,45][3][44][45]. In mice in vivo, glucagon induced tachycardia [65]. In the liver, the desensitization of AC by glucagon has been convincingly demonstrated [109]. Desensitization for all G-protein-coupled receptors involves isoforms of the GTP-binding protein receptor kinase (GRK), protein kinase C, and a cAMP-dependent protein kinase (PKA, [110], Figure 1).
Glucagon can lead to the phosphorylation of the glucagon receptor in Chinese hamster ovary cells transfected with this receptor [111]. One would predict this to occur in the human heart, but this has not yet been reported. For receptor regulation, it is essential to know that glucagon receptors can be ubiquitylated [112,113,114][112][113][114]. This ubiquitination is involved in the internalization of the glucagon receptor upon agonist occupation. In addition, ubiquitination seems to drive biased agonism; it can lead to the stimulation of the non-canonical pathway comprising β-arrestin and MAP kinases [113]. The expression of the glucagon receptor in a transfected cell line could be reduced by antidiabetic drugs, namely thiazolidinediones [115]. One could speculate that this mechanism might be active in the heart. This is consistent with the information available for the promoter of the glucagon receptor, which contains an element involved in the mechanism of action of thiazolidinediones [116]. In the rat glucagon receptor gene, a glucose response element reduces the transcription of the glucagon receptor and, thus, its expression [117]. The promoter region of the human glucagon receptor has been studied [116]. This is relevant for the downregulation of the glucagon receptor. There is evidence for a cAMP-mediated downregulation of promoter transcription in cell culture studies [116]. This would, for instance, explain why glucagon can reduce the expression of the glucagon receptor by raising the cAMP level. This is a protective mechanism against deleterious increases in the cellular (in our case, cardiac) cAMP levels by auto-inhibiting the action of glucagon. Similarly, glucose autoinhibition exists. In liver cells in culture, high glucose concentrations reduce the efficacy of the glucagon receptors to increase glucose concentrations further [100]. Whether this holds true in the heart remains to be studied.
4. Glucagon Receptor Agonists and AntagonistsAs already mentioned above, glucagon has about 140 times less affinity for glucagon-like peptide receptors (about 130 nM) than specific agonists at these receptors (review: [9]). GLP-1-receptor expression as mRNA is three times more abundant in the human atrium than the human ventricle or cardiomyocytes from the left ventricle [94].
Here, species differences occur again, making it challenging to translate animal data directly to patients. In rat cardiomyocytes, glucagon-like peptide-1 increased cAMP levels but reduced contractility. This was accompanied by a reduced pH in the cardiomyocytes and a subsequent desensitization of the myofilament to calcium [118] (review: [119]). In contrast, the stimulation of GLP-1 receptors increased the force of contraction in human atrial preparations [94]. Peptides similar to glucagon can be used as agonists and antagonists at the glucagon receptors [119,120][119][120]. Taking a pharmacokinetic approach, non-peptide antagonists at the glucagon receptor have been developed that are per-orally available and might play a role in treating diabetes [12,119][12][119]. Some authors have claimed that glucagon fails to pass through the blood–brain barrier [121]. Hence, glucagon infused to treat the heart should not have direct side effects in the brain. However, this view has been challenged in recent years; some have claimed that glucagon can pass into the brain and exert physiological effects in the brain [122].
There has been some success in devising drugs comprising glucagon, a linker, and triiodothyronine. This bifunctional agonist was helpful in a mouse model of diabetes for preserving cardiovascular function [123]. Peptide fragments of glucagon or mutated glucagon stimulate the activity of AC in a cell culture system, usually with a potency and effectivity less than those of native glucagon. As such, these derivatives of glucagon are partial agonists, and thus, they are also antagonists at the glucagon receptor [82]. Another classification of glucagon receptor antagonists is to divide them based on their chemistry into small organic molecules (not protease-sensitive peptides), antibodies, or antisense ribonucleic acid (RNA) [124]. Theoretically, antisense RNA could also be encapsulated in a virus. Antagonists were initially designed to reduce the blood glucose levels (mice and monkeys: [125]).
Peptide analogs of glucagon have been radioactively labeled and used as tracers to study the glucagon receptor occupation in the living body [126]. Like insulin, glucagon can precipitate in solution or stick to the vessel wall. Hence, some have recommended the addition of bovine serum albumin and an acidic pH for its storage [127]. Others have developed a mutated glucagon called dasiglucagon, which contains seven amino acid mutations compared to glucagon and does not cause fibrils to form in aqueous solutions [113]. |
References
- Ahrén, B. Glucagon—Early breakthroughs and recent discoveries. Peptides 2015, 67, 74–81.
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091.
- Farah, A.; Tuttle, R. Studies on the pharmacology of glucagon. J. Pharmacol. Exp. Ther. 1960, 129, 49–55.
- Lucchesi, B.R. Cardiac actions of glucagon. Circ. Res. 1968, 22, 777–787.
- Glick, G.; Parmley, W.W.; Wechsler, A.S.; Sonnenblick, E.H. Glucagon. Its enhancement of cardiac performance in the cat and dog and persistence of its inotropic action despite beta-receptor blockade with propranolol. Circ. Res. 1968, 22, 789–799.
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439.
- Baiio, L.L.; Yusta, B.; Mulvihill, E.E.; Cao, X.; Streutker, C.J.; Butany, J.; Cappola, T.P.; Margulies, K.B.; Drucker, D.J. GLP 1 receptor expression within the human heart. Endocrinology 2018, 159, 1570–1584.
- Munroe, D.G.; Gupta, A.K.; Kooshesh, F.; Vyas, T.B.; Rizkalla, G.; Wang, H.; Demchyshyn, L.; Yang, Z.J.; Kamboj, R.K.; Chen, H. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1999, 96, 1569–1573.
- Jelinek, L.J.; Lok, S.; Rosenberg, G.B.; Smith, R.A.; Grant, F.J.; Biggs, S.; Bensch, P.A.; Kuijper, J.L.; Sheppard, P.O.; Sprecher, C.A.; et al. Expression cloning and signaling properties of the rat glucagon receptor. Science 1993, 259, 1614–1616.
- MacNeil, D.J.; Occi, J.L.; Hey, P.J.; Strader, C.D.; Graziano, M.P. Cloning and expression of a human glucagon receptor. Biochem. Biophys. Res. Commun. 1994, 198, 328–334.
- Koth, C.M.; Murray, J.M.; Mukund, S.; Madjidi, A.; Minn, A.; Clarke, H.J.; Wong, T.; Chiang, V.; Luis, E.; Estevez, A.; et al. Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 14393–14398.
- Yang, D.H.; Zhou, C.H.; Liu, Q.; Wang, M.W. Landmark studies on the glucagon subfamily of GPCRs: From small molecule modulators to a crystal structure. Acta Pharmacol. Sin. 2015, 36, 1033–1042.
- Yang, L.; Yang, D.; de Graaf, C.; Moeller, A.; West, G.M.; Dharmarajan, V.; Wang, C.; Siu, F.Y.; Song, G.; Reedtz-Runge, S. Conformational states of the full-length glucagon receptor. Nat. Commun. 2015, 6, 7859.
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110.
- Krishna Kumar, K.; O’Brien, E.S.; Habrian, C.H.; Latorraca, N.R.; Wang, H.; Tuneew, I.; Montabana, E.; Marqusee, S.; Hilger, D.; Isacoff, E.Y.; et al. Negative allosteric modulation of the glucagon receptor by RAMP2. Cell 2023, 186, 1465–1477.e18.
- Svoboda, M.; Tastenoy, M.; Vertongen, P.; Robberecht, P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell. Endocrinol. 1994, 105, 131–137.
- Hansen, L.H.; Abrahamsen, N.; Nishimura, E. Glucagon receptor mRNA distribution in rat tissues. Peptides 1995, 16, 1163–1166.
- Ali, S.; Ussher, J.R.; Baggio, L.L.; Kabir, M.G.; Charron, M.J.; Ilkayeva, O.; Newgard, C.B.; Drucker, D.J. Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Mol. Metab. 2014, 4, 132–143.
- Baggio, L.L.; Ussher, J.R.; McLean, B.A.; Cao, X.; Kabir, M.G.; Mulvihill, E.E.; Mighiu, A.S.; Zhang, H.; Ludwig, A.; Seeley, R.J.; et al. The autonomic nervous system and cardiac GLP-1 receptors control heart rate in mice. Mol. Metab. 2017, 6, 1339–1349.
- Watanabe, M.; Hirose, Y.; Sugimoto, M.; Nakanishi, M.; Watanabe, H.; Shimada, M. The distribution of tissue insulin receptors in the mouse by whole-body autoradiography. J. Recept. Res. 1992, 12, 13–37.
- Lok, S.; Kuijper, J.L.; Jelinek, L.J.; Kramer, J.M.; Whitmore, T.E.; Sprecher, C.A.; Mathewes, S.; Grant, F.J.; Biggs, S.H.; Rosenberg, G.B.; et al. The human glucagon receptor encoding gene: Structure, cDNA sequence and chromosomal localization. Gene 1994, 140, 203–209.
- Sivarajah, P.; Wheeler, M.B.; Irwin, D.M. Evolution of receptors for proglucagon-derived peptides: Isolation of frog glucagon receptors. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 128, 517–527.
- Aranda-Domene, R.; Orenes-Piñero, E.; Arribas-Leal, J.M.; Canovas-Lopez, S.; Hernández-Cascales, J. Evidence for a lack of inotropic and chronotropic effects of glucagon and glucagon receptors in the human heart. Cardiovasc. Diabetol. 2023, 22, 128.
- Bomholt, A.B.; Johansen, C.D.; Christensen, J.B.; Kjeldsen, S.A.S.; Galsgaard, K.D.; Winther-Sørensen, M.; Serizawa, R.; Hornum, M.; Porrini, E.; Pedersen, J.; et al. Evaluation of commercially available glucagon receptor antibodies and glucagon receptor expression. Commun. Biol. 2022, 5, 1278.
- Nobel-Allen, N.; Kirsch, M.; Lucchesi, B.R. Glucagon: Its enhancement of cardiac performance in the cat with chronic heart failure. J. Pharmacol. Exp. Ther. 1973, 187, 475–481.
- Sattler, R.W.; van Zwieten, P.A. The positive inotropic action of glucagon on the cat heart in situ. Klin. Wochenschr. 1972, 50, 531–533.
- Winokur, S.; Nobel-Allen, N.L.; Lucchesi, B.R. The positive inotropic effect of glucagon in the chronically failed right ventricle as demonstrated in the isolated cat heart. Eur. J. Pharmacol. 1975, 32, 349–356.
- Gold, H.K.; Prindle, K.H.; Levey, G.S.; Epstein, S.E. Effects of experimental heart failure on the capacity of glucagon to augment myocardial contractility and activate adenyl cyclase. J. Clin. Investig. 1970, 49, 999–1006.
- Marcus, M.L.; Skelton, C.L.; Prindle, K.H., Jr.; Epstein, S.E. Potentiation of the inotropic effects of glucagon by theophylline. J. Pharmacol. Exp. Ther. 1971, 179, 331–337.
- Regan, T.J.; Lehan, P.H.; Henneman, D.H.; Behar, A.; Hellems, H.K. Myocardial metabolic and contractile response to glucagon and Epinephrine. J. Lab. Clin. Med. 1964, 63, 638–647.
- Endoh, M. Correlation of cyclic AMP and cyclic GMP levels with changes in contractile force of dog ventricular myocardium during cholinergic antagonism of positive inotropic actions of histamine, glucagon, theophylline and papaverine. JPN J. Pharmacol. 1979, 29, 855–864.
- Antonaccio, M.J.; Lucchesi, B.R. The interaction of glucagon with theophylline, PGE1, isoproteerenol, ouabain, and CaC1 on the dog isolated papillary muscle. Life Sci. 1970, 9, 1081–1089.
- Nayler, W.G.; McInnes, I.; Chipperfield, D.; Carson, V.; Daile, P. The effect of glucagon on calcium exchangeability, coronary blood flow, myocardial function and high energy phosphate stores. J. Pharmacol. Exp. Ther. 1970, 171, 265–275.
- Lipski, J.I.; Kaminsky, D.; Donoso, E.; Friedberg, C.K. Electrophysiological effects of glucagon on the normal canine heart. Am. J. Physiol. 1972, 222, 1107–1112.
- Furukawa, Y.; Saegusa, K.; Ogiwara, Y.; Chiba, S. Different effectiveness of glucagon on the pacemaker activity and contractility in intact dog hearts and in isolated perfused right atria. JPN Heart J. 1986, 27, 215–222.
- Chiba, S. Positive chronotropic and inotropic effects of glucagon on the canine isolated atrium. Tohoku J. Exp. Med. 1975, 115, 61–65.
- Kimura, T.; Kokubun, M.; Hashimoto, K. Primary effect of glucagon on positive chronotropism. Jpn. J. Pharmacol. 1974, 24, 279–283.
- Smitherman, T.C.; Osborn, R.C., Jr.; Atkins, J.M. Cardiac dose response relationship for intravenously infused glucagon in normal intact dogs and men. Am. Heart J. 1978, 96, 363–371.
- Bache, R.J.; McHale, P.A.; Curry, C.L.; Alexander, J.A.; Greenfield, J.C., Jr. Coronary and systemic hemodynamic effects of glucagon in the intact unanesthetized dog. J. Appl. Physiol. 1970, 29, 769–774.
- Moir, T.W.; Naylor, W.G. Coronary vascular effects of glucagon in the isolated dog heart. Circ. Res. 1970, 26, 29–34.
- Iwanij, V.; Hur, K.C. Development of physiological responsiveness to glucagon during embryogenesis of avian heart. Dev. Biol. 1987, 122, 146–152.
- Méry, P.F.; Brechler, V.; Pavoine, C.; Pecker, F.; Fischmeister, R. Glucagon stimulates the cardiac Ca2+ current by activation of adenylyl cyclase and inhibition of phosphodiesterase. Nature 1990, 345, 158–161.
- Yao, L.F.; MacLeod, K.M.; McNeill, J.H. Glucagon-induced densensitization: Correlation between cyclic AMP levels and contractile force. Eur. J. Pharmacol. 1982, 79, 147–150.
- MacLeod, K.M.; Rodgers, R.L.; McNeill, J.H. Characterization of glucagon-induced changes in rate, contractility and cyclic AMP levels in isolated cardiac preparations of the rat and guinea pig. J. Pharmacol. Exp. Ther. 1981, 217, 798–804.
- Rodgers, R.L.; MacLeod, K.M.; McNeill, J.H. Responses of rat and guinea pig hearts to glucagon. Lack of evidence for a dissociation between changes in myocardial cyclic 3′5′-adenosine monophosphate and contractility. Circ. Res. 1981, 49, 216–225.
- Hadházy, P. Actions of glucagon and dibutyryl cyclic 3′,5′-AMP on chronotropic responses to vagal stimulation and acetylcholine. Pharmacology 1973, 9, 285–293.
- Green, R.L. Paradoxical effects of calcium-glucagon interaction on cardiac muscle contractility of isolated guinea pig atria. Pharmacology 1977, 15, 519–528.
- Spilker, B. Comparison of the inotropic response to glucagon, ouabain and noradrenaline. Br. J. Pharmacol. 1970, 40, 382–395.
- Klein, S.W.; Morch, J.E.; Mahon, W.A. Cardiovascular effects of glucagon in man. Can. Med. Assoc. J. 1968, 98, 1161–1164.
- Strauer, B.E. The influence of glucagon on myocardial mechanics of papillary muscles obtained from patients with chronic congestive heart failure. Naunyn Schmiedebergs Arch. Pharmakol. 1971, 270, 90–93.
- Linhart, J.W.; Barold, S.S.; Cohen, L.S.; Hildner, F.J.; Samet, P. Cardiovascular effects of glucagon in man. Am. J. Cardiol. 1968, 22, 706–710.
- Williams, J.F., Jr. Glucagon and the cardiovascular system. Ann. Intern. Med. 1969, 71, 419–423.
- Manchester, J.H.; Parmley, W.W.; Matloff, J.M.; Leidtke, A.J.; LaRaia, P.J.; Herman, M.V.; Sonnenblock, E.H.; Gorlin, R. Effects of glucagon on myocardial oxygen consumption and coronary blood flow in man and in dog. Circulation 1970, 41, 579–588.
- Wildenthal, K.; Allen, D.O.; Karlsson, J.; Wakeland, J.R.; Clark, C.M., Jr. Responsiveness to glucagon in fetal hearts. Species variability and apparent disparities between changes in beating, adenylate cyclase activation, and cyclic AMP concentration. J. Clin. Investig. 1976, 57, 551–558.
- Boder, G.B.; Harley, R.J.; Johnson, I.S. Recording system for monitoring automaticity of heart cells in culture. Nature 1971, 231, 531–532.
- Boder, G.B.; Johnson, I.S. Comparative effects of some cardioactive agents on automaticity of cultured heart cells. J. Mol. Cell. Cardiol. 1972, 4, 453–463.
- Necco, A.; Dasdia, T.; Di Francesco, D.; Ferroni, A. Action of ouabain, oligomycin, and glucagon on cultured heart cells treated with adriamycin. Pharmacol. Res. Commun. 1976, 8, 105–109.
- Greeff, K. Einfluss von Pharmaka auf die Kontraktilität des Herzens . Verh. Dtsch. Ges. Kreislaufforsch. 1976, 42, 80–92. (In German)
- Henry, P.D.; Dobson, J.G., Jr.; Sobel, B.E. Dissociations between changes in myocardial cyclic adenosine monophosphate and contractility. Circ. Res. 1975, 36, 392–400.
- Goldstein, R.E.; Skelton, C.L.; Levey, G.S.; Glancy, D.L.; Beiser, G.D.; Epstein, S.E. Effects of chronic heart failure on the capacity of glucagon to enhance contractility and adenyl cyclase activity of human papillary muscles. Circulation 1971, 44, 638–648.
- Prasad, K. Glucagon-induced changes in the action potential, contraction, and Na+-K+-ATPase of cardiac muscle. Cardiovasc. Res. 1975, 9, 355–365.
- Hernández-Cascales, J. Does glucagon have a positive inotropic effect in the human heart? Cardiovasc. Diabetol. 2018, 17, 148.
- Petersen, K.M.; Bøgevig, S.; Holst, J.J.; Knop, F.K.; Christensen, M.B. Hemodynamic Effects of Glucagon: A Literature Review. J. Clin. Endocrinol. Metab. 2018, 103, 1804–1812.
- Parmley, W.W.; Glick, G.; Sonnenblick, E.H. Cardiovascular effects of glucagon in man. N. Engl. J. Med. 1968, 279, 12–17.
- Mukharji, A.; Drucker, D.J.; Charron, M.J.; Swoap, S.J. Oxyntomodulin increases intrinsic heart rate through the glucagon receptor. Physiol. Rep. 2013, 1, e00112.
- Kiss, Z.; Tkachuk, V.A. Guanine-nucleotide-dependent inhibition of adenylate cyclase of rabbit heart by glucagon. Eur. J. Biochem. 1984, 142, 323–328.
- Gonzalez-Muñoz, C.; Nieto-Cerón, S.; Cabezas-Herrera, J.; Hernández-Cascales, J. Glucagon increases contractility in ventricle but not in atrium of the rat heart. Eur. J. Pharmacol. 2008, 587, 243–247.
- Harney, J.A.; Rodgers, R.L. Insulin-like stimulation of cardiac fuel metabolism by physiological levels of glucagon: Involvement of PI3K but not cAMP. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E155–E161.
- Brunt, M.E.; McNeill, J.H. The effect of glucagon on rat cardiac cyclic AMP, phosphorylase a and force of contraction. Arch. Int. Pharmacodyn. Ther. 1978, 233, 42–52.
- Merino, B.; Quesada, I.; Hernández-Cascales, J. Glucagon Increases Beating Rate but Not Contractility in Rat Right Atrium. Comparison with Isoproterenol. PLoS ONE 2015, 10, e0132884.
- Mayer, S.E.; Namm, D.H.; Rice, L. Effect of glucagon on cyclic 3′,5′-AMP, phosphorylase activity and contractility of heart muscle of the rat. Circ. Res. 1970, 26, 225–233.
- Clark, C.M., Jr.; Beatty, B.; Allen, D.O. Evidence for delayed development of the glucagon receptor of adenylate cyclase in the fetal and neonatal rat heart. J. Clin. Investig. 1973, 52, 1018–1025.
- Graziano, M.P.; Hey, P.J.; Borkowski, D.; Chicchi, G.G.; Strader, C.D. Cloning and functional expression of a human glucagon-like peptide-1 receptor. Biochem. Biophys. Res. Commun. 1993, 196, 141–146.
- Levey, G.S.; Epstein, S.E. Activation of adenyl cyclase by glucagon in cat and human heart. Circ. Res. 1969, 24, 151–156.
- Sauvadet, A.; Rohn, T.; Pecker, F.; Pavoine, C. Synergistic actions of glucagon and miniglucagon on Ca2+ mobilization in cardiac cells. Circ. Res. 1996, 78, 102–109.
- Chatelain, P.; Robberecht, P.; Waelbroeck, M.; De Neef, P.; Camus, J.C.; Huu, A.N.; Roba, J.; Christophe, J. Topographical distribution of the secretin- and VIP-stimulated adenylate cyclase system in the heart of five animal species. Pflug. Arch. 1983, 397, 100–105.
- Brechler, V.; Pavoine, C.; Hanf, R.; Garbarz, E.; Fischmeister, R.; Pecker, F. Inhibition by glucagon of the cGMP-inhibited low-Km cAMP phosphodiesterase in heart is mediated by a pertussis toxin-sensitive G-protein. J. Biol. Chem. 1992, 267, 15496–15501.
- Kilts, J.D.; Gerhardt, M.A.; Richardson, M.D.; Sreeram, G.; Mackensen, G.B.; Grocott, H.P.; White, W.D.; Davis, R.D.; Newman, M.F.; Reves, J.G.; et al. Beta(2)-adrenergic and several other G protein-coupled receptors in human atrial membranes activate both G(s) and G(i). Circ. Res. 2000, 87, 705–709.
- Menon, K.M.; Giese, S.; Jaffe, R.B. Hormone- and fluoride-sensitive adenylate cyclases in human fetal tissues. Biochim. Biophys. Acta 1973, 304, 203–209.
- Brown, H.D.; Chattopadhyay, S.K.; Matthews, W.S. Glucagon stimulation of adenyl cyclase activity of cardiac muscle. Naturwissenschaften 1968, 55, 181–182.
- Christophe, J.; Waelbroeck, M.; Chatelain, P.; Robberecht, P. Heart receptors for VIP, PHI and secretin are able to activate adenylate cyclase and to mediate inotropic and chronotropic effects. Species variations and physiopathology. Peptides 1984, 5, 341–353.
- Robberecht, P.; Damien, C.; Moroder, L.; Coy, D.H.; Wünsch, E.; Christophe, J. Receptor occupancy and adenylate cyclase activation in rat liver and heart membranes by 10 glucagon analogs modified in position 2, 3, 4, 25, 27 and/or 29. Regul. Pept. 1988, 21, 117–128.
- England, P.J. Studies on the phosphorylation of the inhibitory subunit of troponin during modification of contraction in perfused rat heart. Biochem. J. 1976, 160, 295–304.
- Juan-Fita, M.J.; Vargas, M.L.; Kaumann, A.J.; Hernández Cascales, J. Rolipram reduces the inotropic tachyphylaxis of glucagon in rat ventricular myocardium. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 324–329.
- Juan-Fita, M.J.; Vargas, M.L.; Hernández, J. The phosphodiesterase 3 inhibitor cilostamide enhances inotropic responses to glucagon but not to dobutamine in rat ventricular myocardium. Eur. J. Pharmacol. 2005, 512, 207–213.
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ. Res. 2006, 98, 1081–1088.
- Carlson, E.C.; Grosso, D.S.; Romero, S.A.; Frangakis, C.J.; Byus, C.V.; Bressler, R. Ultrastructural studies of metabolically active isolated adult rat heart myocytes. J. Mol. Cell. Cardiol. 1978, 10, 449–459.
- Watanabe, A.M.; Hathaway, D.R.; Besch, H.R., Jr.; Farmer, B.B.; Harris, R.A. alpha-Adrenergic reduction of cyclic adenosine monophosphate concentrations in rat myocardium. Circ. Res. 1977, 40, 596–602.
- Cascieri, M.A.; Koch, G.E.; Ber, E.; Sadowski, S.J.; Louizides, D.; de Laszlo, S.E.; Hacker, C.; Hagmann, W.K.; MacCoss, M.; Chicchi, G.G.; et al. Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J. Biol. Chem. 1999, 274, 8694–8697.
- Murad, F.; Vaughan, M. Effect of glucagon on rat heart adenyl cyclase. Biochem. Pharmacol. 1969, 18, 1053–1059.
- Shiao, L.L.; Cascieri, M.A.; Trumbauer, M.; Chen, H.; Sullivan, K.A. Generation of mice expressing the human glucagon receptor with a direct replacement vector. Transgenic Res. 1999, 8, 295–302.
- Livingston, J.N.; Einarsson, K.; Backman, L.; Ewerth, S.; Arner, P. Glucagon receptor of human liver. Studies of its molecular weight and binding properties, and its ability to activate hepatic adenylyl cyclase of non-obese and obese subjects. J. Clin. Investig. 1985, 75, 397–403.
- Rodgers, R.L. A reappraisal of the role of cyclic AMP in the physiological action of glucagon. Peptides 2023, 159, 170906.
- Wallner, M.; Kolesnik, E.; Ablasser, K.; Khafaga, M.; Wakula, P.; Ljubojevic, S.; Thon-Gutschi, E.M.; Sourij, H.; Kapl, M.; Edmunds, N.J.; et al. Exenatide exerts a PKA-dependent positive inotropic effect in human atrial myocardium: GLP-1R mediated effects in human myocardium. J. Mol. Cell. Cardiol. 2015, 89 Pt B, 365–375.
- Huh, K.Y.; Hwang, J.G.; Shin, W.; Baek, S.; Choi, J.; Lee, N.; Cho, Y.M.; Lee, H. A double-blind, placebo-controlled, single-ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of HM15136, a novel long-acting glucagon analogue, in healthy subjects. Diabetes Obes. Metab. 2022, 24, 411–420.
- Qureshi, S.A.; Rios Candelore, M.; Xie, D.; Yang, X.; Tota, L.M.; Ding, V.D.; Li, Z.; Bansal, A.; Miller, C.; Cohen, S.M.; et al. A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes 2004, 53, 3267–3273.
- O’Harte, F.P.; Franklin, Z.J.; Rafferty, E.P.; Irwin, N. Characterisation of structurally modified analogues of glucagon as potential glucagon receptor antagonists. Mol. Cell. Endocrinol. 2013, 381, 26–34.
- Chepurny, O.G.; Matsoukas, M.T.; Liapakis, G.; Leech, C.A.; Milliken, B.T.; Doyle, R.P.; Holz, G.G. Nonconventional glucagon and GLP-1 receptor agonist and antagonist interplay at the GLP-1 receptor revealed in high-throughput FRET assays for cAMP. J. Biol. Chem. 2019, 294, 3514–3531, Erratum in J. Biol. Chem. 2019, 294, 8714.
- Irwin, N.; Franklin, Z.J.; O’Harte, F.P. desHis1Glu9-glucagon- and desHis1Glu9(Lys30PAL)-glucagon: Long-acting peptide-based PEGylated and acylated glucagon receptor antagonists with potential antidiabetic activity. Eur. J. Pharmacol. 2013, 709, 43–51.
- Gao, J.; Li, H.; Xu, H.; Liu, Y.; Cai, M.; Shi, Y.; Zhang, J.; Wang, H. High glucose-induced glucagon resistance and membrane distribution of GCGR revealed by super-resolution imaging. iScience 2023, 26, 105967.
- Kjeldsen, S.A.S.; Hansen, L.H.; Esser, N.; Mongovin, S.; Winther-Sørensen, M.; Galsgaard, K.D.; Hunt, J.E.; Kissow, H.; Ceutz, F.R.; Terzic, D.; et al. Neprilysin Inhibition Increases Glucagon Levels in Humans and Mice with Potential Effects on Amino Acid Metabolism. J. Endocr. Soc. 2021, 5, bvab084.
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Gaarde, W.A.; Reed, C.; She, P.; Jetton, T.L.; Demarest, K.T. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 2004, 53, 410–417.
- van Dongen, M.G.; Geerts, B.F.; Morgan, E.S.; Brandt, T.A.; de Kam, M.L.; Romijn, J.A.; Cohen, A.F.; Bhanot, S.; Burggraaf, J. First proof of pharmacology in humans of a novel glucagon receptor antisense drug. J. Clin. Pharmacol. 2015, 55, 298–306.
- Abrahamsen, N.; Lundgren, K.; Nishimura, E. Regulation of glucagon receptor mRNA in cultured primary rat hepatocytes by glucose and cAMP. J. Biol. Chem. 1995, 270, 15853–15857.
- Robberecht, P.; Pochet, R.; Chatelain, P.; Verloes, A.; Camus, J.C.; de Neef, P.; Christophe, J. Decreased secretin and glucagon responsiveness of adenylate cyclase in cardiac membranes from hypothyroid rats. FEBS Lett. 1981, 132, 33–36.
- Chatelain, P.; Robberecht, P.; De Neef, P.; Camus, J.C.; Christophe, J. Early decrease in secretin-, glucagon-, and isoproterenol-stimulated cardiac adenylate cyclase activity in rats treated with isoproterenol. Biochem. Pharmacol. 1982, 31, 347–352.
- Taton, G.; Chatelain, P.; Delhaye, M.; Camus, J.C.; De Neef, P.; Waelbroeck, M.; Tatemoto, K.; Robberecht, P.; Christophe, J. Vasoactive intestinal peptide (VIP) and peptide having N-terminal histidine and C-terminal isoleucine amide (PHI) stimulate adenylate cyclase activity in human heart membranes. Peptides 1982, 3, 897–900.
- Srikant, C.B.; Freeman, D.; McCorkle, K.; Unger, R.H. Binding and biologic activity of glucagon in liver cell membranes of chronically hyperglucagonemic rats. J. Biol. Chem. 1977, 252, 7434–7438.
- Iyengar, R.; Mintz, P.W.; Swartz, T.L.; Birnbaumer, L. Divalent cation-induced desensitization of glucagon-stimulable adenylyl cyclase in rat liver plasma membrane. GTP-dependent stimulation by glucagon. J. Biol. Chem. 1980, 255, 11875–11882.
- Krilov, L.; Nguyen, A.; Miyazaki, T.; Unson, C.G.; Williams, R.; Lee, N.H.; Ceryak, S.; Bouscarel, B. Dual mode of glucagon receptor internalization: Role of PKCα, GRKs and β-arrestins. Exp. Cell Res. 2011, 317, 2981–2994.
- Heurich, R.O.; Buggy, J.J.; Vandenberg, M.T.; Rossomando, A.J. Glucagon induces a rapid and sustained phosphorylation of the human glucagon receptor in Chinese hamster ovary cells. Biochem. Biophys. Res. Commun. 1996, 220, 905–910.
- Kaur, S.; Chen, Y.; Shenoy, S.K. Agonist-activated glucagon receptors are deubiquitinated at early endosomes by two distinct deubiquitinases to facilitate Rab4a-dependent recycling. J. Biol. Chem. 2020, 295, 16630–16642.
- Kaur, J.; Seaquist, E.R. Translational aspects of glucagon: Current use and future prospects. J. Endocrinol. 2023, 257, e220278.
- Kaur, S.; Sokrat, B.; Capozzi, M.E.; El, K.; Bai, Y.; Jazic, A.; Han, B.; Krishnakumar, K.; D’Alessio, D.A.; Campbell, J.E.; et al. The Ubiquitination Status of the Glucagon Receptor determines Signal Bias. J. Biol. Chem. 2023, 299, 104690.
- Schinner, S.; Dellas, C.; Schroder, M.; Heinlein, C.A.; Chang, C.; Fischer, J.; Knepel, W. Repression of glucagon gene transcription by peroxisome proliferator-activated receptor gamma through inhibition of Pax6 transcriptional activity. J. Biol. Chem. 2002, 277, 1941–1948.
- Mortensen, O.H.; Dichmann, D.S.; Abrahamsen, N.; Grunnet, N.; Nishimura, E. Identification of a novel human glucagon receptor promoter: Regulation by cAMP and PGC-1alpha. Gene 2007, 393, 127–136.
- Iizuka, K.; Tomita, R.; Takeda, J.; Horikawa, Y. Rat glucagon receptor mRNA is directly regulated by glucose through transactivation of the carbohydrate response element binding protein. Biochem. Biophys. Res. Commun. 2012, 417, 1107–1112.
- Vila Petroff, M.G.; Egan, J.M.; Wang, X.; Sollott, S.J. Glucagon-like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ. Res. 2001, 89, 445–452.
- Mayo, K.E.; Miller, L.J.; Bataille, D.; Dalle, S.; Göke, B.; Thorens, B.; Drucker, D.J. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003, 55, 167–194.
- Hawkes, C.P.; De Leon, D.D.; Rickels, M.R. Novel Preparations of Glucagon for the Prevention and Treatment of Hypoglycemia. Curr. Diab. Rep. 2019, 19, 97.
- Watanabe, M.; Tamayama, T.; Hayasaki, H.; Kuno, M.; Kuroda, E.; Watanabe, Y.; Shimada, M. Whole body radioautographic analysis of the in vivo distribution of glucagon receptors in mice. Acta Histochem. Cytochem. 1997, 30, 471–476.
- Abraham, M.A.; Lam, T.K.T. Glucagon action in the brain. Diabetologia 2016, 59, 1367–1371.
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Müller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell 2016, 167, 843–857.e14.
- Wewer Albrechtsen, N.J. Glucagon receptor signaling in metabolic diseases. Peptides 2018, 100, 42–47.
- Yan, H.; Gu, W.; Yang, J.; Bi, V.; Shen, Y.; Lee, E.; Winters, K.A.; Komorowski, R.; Zhang, C.; Patel, J.J.; et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J. Pharmacol. Exp. Ther. 2009, 329, 102–111.
- Velikyan, I.; Haack, T.; Bossart, M.; Evers, A.; Laitinen, I.; Larsen, P.; Plettenburg, O.; Johansson, L.; Pierrou, S.; Wagner, M.; et al. First-in-class positron emission tomography tracer for the glucagon receptor. EJNMMI Res. 2019, 9, 17.
- Rodgers, R.L. Glucagon, cyclic AMP, and hepatic glucose mobilization: A half-century of uncertainty. Physiol. Rep. 2022, 10, e15263.
More