Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Atul Pandey.
Antibiotic resistance in Pseudomonas aeruginosa remains one of the most challenging phenomena of everyday medical science. The universal spread of high-risk clones of multidrug-resistant/extensively drug-resistant (MDR/XDR) clinical P. aeruginosa has become a public health threat. The P. aeruginosa bacteria exhibits remarkable genome plasticity that utilizes highly acquired and intrinsic resistance mechanisms to counter most antibiotic challenges. In addition, the adaptive antibiotic resistance of P. aeruginosa, including biofilm-mediated resistance and the formation of multidrug-tolerant persisted cells, are accountable for recalcitrance and relapse of infections.
- drug-resistant
- public health concern
- antibiotic stewardship
1. Introduction
There are several reasons why multidrug-resistant (MDR) and extensively drug-resistant (XDR) P. aeruginosa strains represent a worldwide health threat. First, P. aeruginosa is extremely opportunistic and can cause serious infections in hospital settings, especially in immunocompromised people. Second, it has versatility in its antibiotic resistance and may transmit drug resistance through multiple routes [1]. High-risk clones are spreading worldwide, posing a public health issue that must be examined and addressed [2]. In 2017, the World Health Organization placed carbapenem-resistant P. aeruginosa in the “critical” group, for which new medicines are needed [3].
2. Inherent Resistome
It is intriguing how P. aeruginosa possesses a peculiar assortment of drug-resistance mechanisms, such as multiple chromosomal-associated genes that confer resistance to antibiotics, as well as complex regulatory pathways involved in both inherent and adaptive resistance [9,10,11,12][4][5][6][7]. When compared to other Gram-negative bacteria, the development of inherent resistance in P. aeruginosa is primarily influenced by the expression of inducible AmpC cephalosporinase, the synthesis of constitutive and inducible efflux pumps, and the low permeability of its outer membrane. The synthesis of inducible beta-lactamase is of utmost importance in the inherent resistance of P. aeruginosa to aminopenicillins and cephalosporins, specifically cefoxitin. These antibiotics are potent inducers of AmpC expression, resulting in an overabundance of cephalosporinase [13][8]. The hydrolytic stability of Imipenem is slightly affected by its strong ability to induce enzymes. The expression of inducible AmpC is important in reducing the natural sensitivity of P. aeruginosa [14][9]. Additionally, the recently discovered imipenemase (IMP) PA5542 may also influence the inherent susceptibility of β-lactam antibiotics [15][10]. Nevertheless, additional research is needed to explore their involvement in innate or acquired resistance. The expression of efflux pump is crucial in reducing the inherent susceptibility to a wide range of β-lactam antibiotics and fluoroquinolones. In a similar manner, the inducible expression of Hyperproduction of efflux-mediated (MexXY) genes exerts a notable impact on the inherent, minimal resistance to aminoglycosides [16][11]. These efflux pump systems effectively expel various classes of antibiotics from the bacterial cell, thereby providing it with intrinsic resistance.
3. Mutational Resistome
In addition to its wide-ranging innate resistance repertoire, P. aeruginosa demonstrates remarkable proficiency in acquiring chromosomal mutations, leading to the emergence of novel antimicrobial-resistant superbugs [1], as summarized in Table 1. The β-lactam resistance mechanism driven by mutation has been observed in 20% of P. aeruginosa [12,17,18][7][12][13]. The deactivation of penicillin binding protein-4 (PBP-4) triggers the activation of the CreBC/BlrAB two-component system, which is responsive to carbon sources and contributes to β-lactam resistance. This activation leads to an additional increase in resistance levels [18][13]. The clinical strains were found to possess distinct mutations that affected the transcriptional regulator of AmpR, a protein responsible for regulating the overexpression of ampC and conferring resistance to beta-lactam antibiotics. The mutations under consideration encompass the R154H mutation, which is associated with the epidemic MDR/XDR ST175 high-risk clone, and the D135N mutation, observed in species other than P. aeruginosa [12][7]. Numerous genetic variations have been documented to enhance the amplification of ampC in various genetic sequences, encompassing those responsible for other amidases (AmpDh2/AmpDh3), PBP5 or PBP7, lytic transglycosylases (MltB and SltB1), MPL (UDP-N-acetylmuramate: L-alanyl—D-glutamyl-Meso-(NADH dehydrogenase I chain N) [13][8].
Table 1.
Key genes that are known to be associated in
| Responsible Genes | Mechanism of Resistance | Associated Antibiotics |
|---|---|---|
| gyrA | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| gyrB | Target modification of Quinolones (DNA gyrase) | Fluoroquinolones |
| parC | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| parE | Target modification of Quinolones (DNA topoisomerase IV) | Fluoroquinolones |
| phoQ, cprS, colR, colS, pmrA, pmrB | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| parR | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexXY) | Aminoglycosides, cefepime | |
| parS | Modification of Lipopolysaccharide (addition, 4-amino-4-deoxy-L-arabinose moiety to the lipid A portion) | Polymyxins |
| Downregulation of OprD | Imipenem, meropenem | |
| Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones | |
| Hyperproduction of efflux-mediated genes (MexXY) | Fluoroquinolones | |
| mexR | Hyperproduction of efflux-mediated genes (MexAB-OprM) | Fluoroquinolones |
| nfxB | Hyperproduction of efflux-mediated genes (MexCD-OprJ) | Fluoroquinolones, cefepime |
| mexS | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| Downregulation of OprD | Imipenem, meropenem | |
| mexT | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| cmrA, mvaT, PA3271 | Hyperproduction of efflux-mediated genes (MexEF-OprN) | Fluoroquinolones |
| mexZ, PA5471.1, amgS | Hyperproduction of efflux-mediated genes (MexXY) | MexXY hyperproduction |
| oprD | Inactivation of Porin channels | Imipenem, meropenem |
| ampD, ampDh2, ampDh3, ampR, dacB, mpl | Hyperproduction of AmpC | Ceftazidime, cefepime, piperacillin–tazobactam |
| ftsI | Target modification (PBP3) | Ceftazidime, cefepime, piperacillin–tazobactam |
| fusA1 | Target modification of Aminoglycoside (elongation factor G) | Aminoglycosides |
| glpT | Transporter protein inactivation GlpT | Fosfomycin |
| rpoB | Rifampin target modification, RNA polymerase β-chain | Rifampin |
Findings from studies have demonstrated that genetic variations that modify the structure of AmpC can lead to the development of resistance against β-lactam antibiotics. This resistance includes the newly developed β-lactam–lactamase inhibitor, as well as the combinations of ceftolozane–tazobactam and ceftazidime–avibactam. This is in addition to the phenomenon of AmpC hyperproduction, as previously reported [19,20,21,22][14][15][16][17]. The development of resistance to ceftolozane–tazobactam and ceftazidime–avibactam was observed in a specific group of P. aeruginosa clinical isolates [23][18]. This resistance was linked to various changes in the amino acid composition of AmpC. Recent findings have unveiled the existence of more than 300 distinct variations of cephalosporinase derived from the Pseudomonas genus. Notably, certain variations have been observed to confer increased resistance to ceftolozane–tazobactam and ceftazidime–avibactam. There is a growing body of evidence suggesting that alterations in penicillin-binding proteins (PBPs), specifically mutations in PBP-3, contribute to the development of resistance to β-lactam antibiotics, alongside β-lactamases. Recent data obtained from individuals diagnosed with cystic fibrosis [24,25][19][20], as well as from strains of bacteria causing epidemics [26,27][21][22], and laboratory experiments conducted in controlled environments [28,29][23][24], have provided evidence indicating that specific alterations in penicillin-binding protein-3 play a role in the emergence of resistance to -lactam antibiotics. The R504C/R504H and F533L mutations, located in the domains responsible for stabilizing the -lactam–penicillin-binding protein-3 inactivation complex, have been frequently documented in the scientific literature [30][25]. The presence of inhibitory deletion/insertion sequences within the OprD gene, as well as distant mutations that enhance the activity of efflux pump systems MexEF-OprN or CzcCBA while simultaneously reducing the expression of OprD, can result in the loss of the carbapenem-specific porin—OprD. The inactivation of OprD often leads to resistance against all conventional anti-pseudomonal β-lactams in a synergistic fashion when combined with AmpC overexpression [31][26]. Another pivotal determinant in resistance is the mutational upregulation of one of the four primary efflux pumps in P. aeruginosa [32,33][27][28]. The prevalence of MexAB-OprM and MexXY overexpression in clinical isolates ranges from 10% to 30%, while the overexpression of MexCD-OprJ and MexEF-OprN is less-frequently observed, occurring in approximately 5% of cases. The OprD porin exhibits either inactivity or downregulation, leading to reduced sensitivity to Meropenem and the inducible synthesis of AmpC. The simultaneous upregulation of MexAB-OprM and downregulation of OprD is a significant determinant of Meropenem resistance in clinical strains [34][29].
More than 20% of isolates frequently demonstrate resistance to imipenem, and the majority of these isolates are deficient in OprD [34][29]. The MexAB-OprM efflux pump exhibits the most extensive substrate specificity, and its upregulation due to mutations leads to decreased susceptibility to all -lactams and fluoroquinolones (with the exception of imipenem). The mutation-driven overexpression of MexXY is a common contributing factor in the resistance of clinical strains to cefepime, in addition to its primary role in intrinsic aminoglycoside resistance [35][30]. The hyperproduction of MexEF-OprN is not commonly observed and primarily impacts fluoroquinolone antibiotics. However, mutations in mexT/mexS genes that lead to MexEF-OprN hyperproduction also result in resistance to imipenem by repressing OprD gene expression [36][31]. In spite of exhibiting heightened resistance to various β-lactams and aminoglycosides, the upregulation of MexCD-OprJ, a phenomenon frequently observed in persistent infections, additionally plays a role in conferring resistance to cefepime [37][32].
Fluoroquinolone resistance in P. aeruginosa often arises due to the overexpression of efflux pumps, as well as mutations occurring in type IV topoisomerases (ParC and ParE) and DNA-gyrases (GyrA and GyrB) [38][33]. From a geographical perspective, the prevalence of fluoroquinolone resistance is observed to be the dominant trait, ranging from 30% to 40% in multiple countries. Recent scientific investigations have elucidated that genetic mutations occurring in the fusA1 gene, responsible for encoding the elongation factor G, have the potential to induce resistance to aminoglycoside antibiotics. This resistance mechanism operates in conjunction with the overexpression of the MexXY genes and the acquisition of genetic pathways through horizontal gene transfer. Indeed, empirical evidence has shown that certain mutations in the FusA1 gene can result in resistance to aminoglycoside antibiotics in laboratory settings [39,40][34][35] and in clinical cases of P. aeruginosa infection, especially in individuals with cystic fibrosis [41][36].
In essence, although the occurrence of colistin resistance is still relatively limited (5%), there has been a recent surge, likely due to its heightened utilization as a final option against infections caused by multidrug resistant/extensively drug resistant bacterial strains. The development of colistin resistance frequently occurs due to alterations in the lipid A component of lipopolysaccharide (LPS) following the addition of 4-amino-4-deoxy-L-arabinose [42][37]. The mutations observed are often associated with the regulatory systems PmrAB and PhoPQ, which result in the activation of the arnBCADTEF operon. In recent studies, it has been demonstrated that mutations in the ParRS two-component regulator play a crucial role in driving colistin resistance. These mutations activate the arnBCATEF genes, leading to an MDR profile. Furthermore, they upregulate the MexXY genes while downregulating the OprD gene [11][6]. The ColRS and CprRS systems have also been implicated in the development of polymyxin resistance [43][38].
4. Horizontally Acquired Resistome
Novel antibiotics of the latest generation have been developed with the specific purpose of selectively inhibiting crucial cellular proteins involved in DNA replication and repair, protein synthesis, and the production of components for the cell membrane [44][39]. The primary strategies employed to address acquired resistance involve implementing chemical modifications to preexisting antibiotics. The rate of antibiotic production has experienced a notable decrease in recent years, despite the fact that a considerable number of antibiotics are presently undergoing their third or fourth round of modifications. Furthermore, due to the evolutionary adaptability of bacteria, the efficacy of antibiotic therapy has progressively diminished over a period of time [45][40]. Furthermore, bacteria have developed a complex regulatory evolutionary adaptation by acquiring resistance genes primarily through conjugation and, to a lesser degree, through spontaneous transformation and transduction [46][41]. Despite the perceived insignificance of transformation, recent research suggests that its importance may be greater than previously hypothesized [47][42]. Recent research examined the effectiveness of horizontal gene transfer (HGT) through conjugation and examined the MDR phenotypes of numerous clinical and environmental bacterial strains from various sources. Along with examining the effects of medications and heavy metal (arsenic), conjugation efficiency between clinical and environmental strains was also examined. They discovered that using 2-HDA as a COIN prevented HGT between strains that were obtained in hospitals and those that naturally exist [48][43].
One aspect of mutational resistance that is of significant interest is the transferable type of P. aeruginosa resistance, which occurs relatively frequently and contributes to the overall accumulation of concern. Bacterial conjugation serves as the fundamental mechanism for both intra- and inter-species HGTs. It plays a crucial role in expediting the dissemination of antibiotic resistance genes [49][44]. Certainly, the prevalence of highly hazardous transferable β-lactamases, including ESBLs and carbapenemases (specifically class B carbapenemases, also known as Metallo β-lactamases), is steadily increasing on a global scale. Nevertheless, their distribution exhibits inconsistency and displays variation across hospitals and regions, ranging from less than 1% to approximately 50% [50][45]. Moreover, the occurrence of transferable β-lactamases in P. aeruginosa might have been underestimated in several locations due to the challenges associated with their detection [51][46]. Integrons belonging to Class 1 generally encompass determinants of resistance to aminoglycosides, as well as the genes responsible for extended-spectrum beta-lactamases (ESBLs) and carbapenemases. While the involvement of conjugative elements is now more commonly recognized, these integrons are often inserted into transposable elements located on the bacterial chromosome [51,52,53,54][46][47][48][49]. A recent study was conducted to review the distribution of spreadable β-lactamases in P. aeruginosa [55][50]. Frequently documented extended-spectrum beta-lactamases (ESBLs) in P. aeruginosa encompass class D enzymes, specifically OXA-2 or OXA-10 variants, as well as class A enzymes including PER, VEB, GES, BEL, and PME variants. Metallo β-lactamases (MBLs) are the predominant carbapenemases found in P. aeruginosa. Among these MBLs, the VIM and IMP variants are the most prevalent and widely distributed across different geographical regions. In Brazil, the prevalence of the SPM MBL gene is extensive, while the NDM, GIM, and FIM genes are sporadically detected. The prevalence of Class A carbapenemases in P. aeruginosa is relatively low on a global scale, even though GES and KPC enzymes have been identified in multiple countries [54][49].
The transferability resistance of aminoglycosides is influenced by the presence of aminoglycoside-modifying enzymes that are encoded within Class 1 integrons. The acetyltransferases frequently observed in P. aeruginosa are AAC 3′ gentamicin and AAC 6′ tobramycin, as well as the nucleotidyltransferase ANT 2′-I gentamicin and tobramycin. Nevertheless, there are significant emerging concerns associated with 16S rRNA methyltransferases, such as Rmt or Arm, as they confer resistance to all commercially available aminoglycosides, including the recently developed plazomicin [54][49]. Intermittently, it has been observed that the prevalence of transferable resistance to fluoroquinolones is primarily influenced by Qnr determinants, such as QnrVC1 [56][51]. In a recent scientific study, it has been demonstrated that a novel phosphotransferase, known as CrpP, is responsible for facilitating plasmid-mediated quinolone resistance [57][52].
Ceftolozane–tazobactam and Ceftazidime–avibactam, two recently developed combinations, exhibit a notable degree of resistance to AmpC hydrolysis [58,59][53][54]. This resistance is attributed to the inhibitory effect of avibactam on AmpC in the case of ceftazidime–avibactam, and the ability of ceftolozane to remain stable against hydrolysis by AmpC in the case of ceftolozane–tazobactam. Nevertheless, based on existing in vitro and in vivo research, it appears that the emergence of resistance to both drugs could be attributed to a combination of genetic mutations, leading to increased production of AmpC and alterations in its structure [19,20,22][14][15][17]. The empirical evidence obtained from experiments conducted in living organisms (in vivo) and in controlled laboratory conditions (in vitro) suggests that particular genetic alterations in penicillin-binding protein-3 have the potential to reduce the vulnerability to the aforementioned combinations. The susceptibility of ceftazidime–avibactam seems to be more influenced by the overexpression of different efflux pumps compared to ceftolozane–tazobactam [23,60][18][55]. Both ceftolozane–tazobactam and ceftazidime–avibactam have demonstrated a lack of efficacy against strains that produce acquired β-lactamases. Ceftazidime–avibactam, but not ceftolozane–tazobactam, exhibits potential activity against isolates that generate class A carbapenemases, such as GES enzymes [61][56]. In a similar vein, the effectiveness of ceftolozane–tazobactam and ceftazidime–avibactam against strains of P. aeruginosa that produce extended-spectrum beta-lactamase (ESBL) exhibits variability, albeit with a generally favorable outcome for ceftazidime–avibactam. Ultimately, the development of resistance to both pharmaceutical agents can arise due to the presence of extended-spectrum mutations in horizontally transferred OXA-type β-lactamases [62,63][57][58].
5. Antibiotic Resistance by SOS Response
A universally preserved bacterial stress response is primarily triggered by DNA damage. The SOS response initiates and coordinates various biological processes, including DNA repair mechanisms, bacterial cell division arrest, and latent bacteriophage induction. The SOS response is characterized by the activation of DNA polymerases IV and V. This occurs when the DNA damage is prolonged and significant. (Figure 1) depicts the visual representation of the data or information being discussed. Bacterial cultures cultivated in an artificial environment were employed in nearly all studies pertaining to the SOS response and the development of resistance to antibiotics. Prior studies have established a correlation between the mutator phenotype and the ability of bacteria, such as P. aeruginosa, to cause chronic infections in individuals with cystic fibrosis [64,65][59][60]. Research examining the genetic alterations in consecutively recovered strains has provided evidence supporting the activation of the SOS response in vivo [66][61]. Additionally, other indirect evidence has also been reported [67,68][62][63].
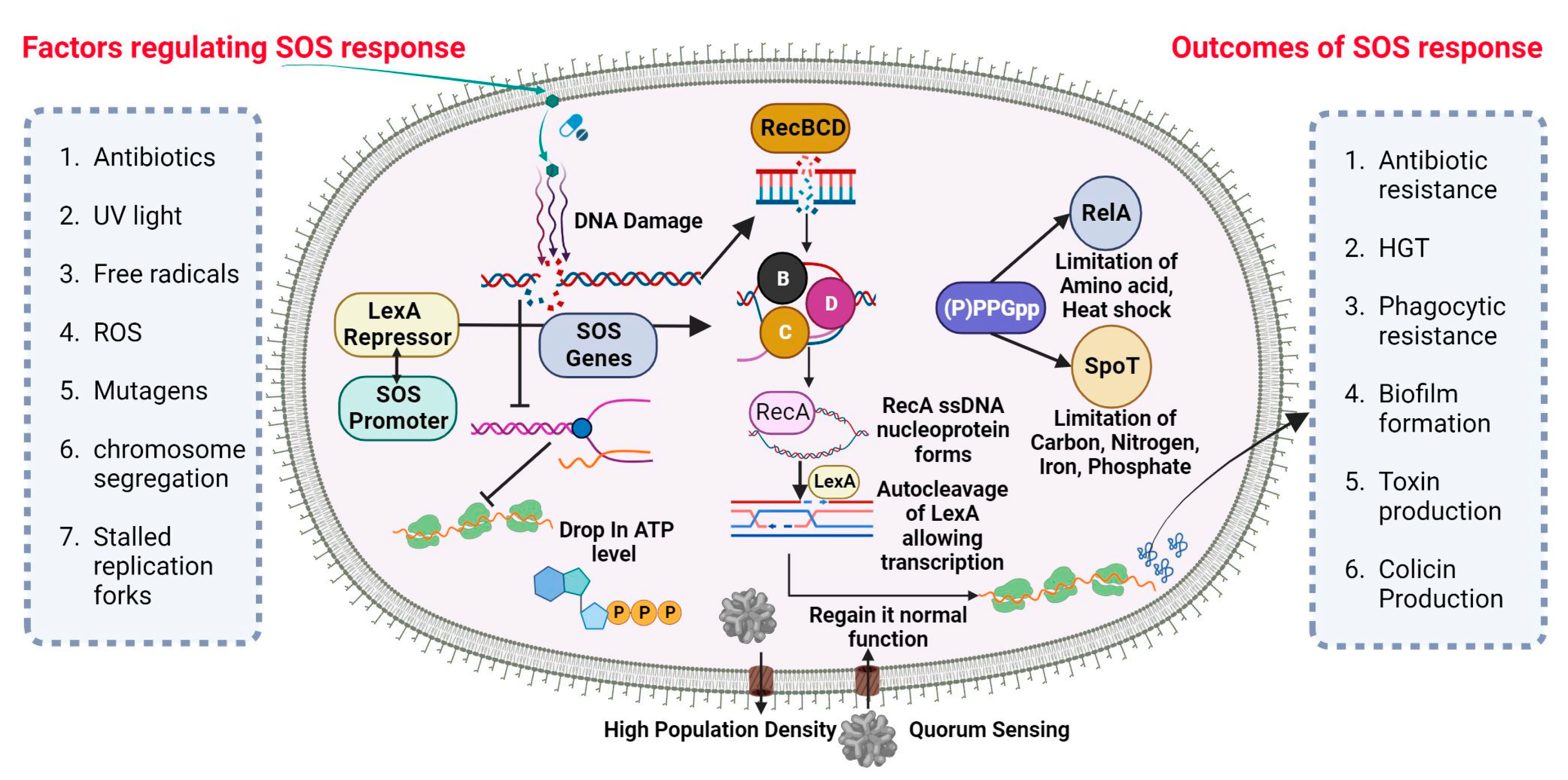
Figure 1. SOS DNA repair mechanism in P. aeruginosa. Nucleic acid inhibitor antibiotic nitration can stimulate the SOS response in P. aeruginosa via the synthesis of the RecA gene from RecBCD subunit and upregulating Lex-containing TisAB, leading to an ATP level drop and the downregulation of important cellular functions. RecA filaments merge and trigger the SOS response. When LexA and RecA-ssDNA nucleoprotein filament connect, LexA’s latent protease activity is activated, leading to LexA’s autocleavage. After LexA is autocleaved and rendered inactive, the SOS gene’s transcription is triggered, inducing a global transcriptional response.
SOS-Dependent Mutagenesis and Resistance
The cellular SOS response is a captivating bacterial defense mechanism through which bacteria can acquire drug resistance, induce mutagenesis, and undergo genome reorganization [69][64]. The Lexi promoter-binding repressor protein regulates the SOS system. Upon binding to the RecA filament, the LexA protein undergoes self-cleavage, resulting in a reduction in LexA protein levels within the cell. This process subsequently triggers the activation of over 40 genes in bacterial cells, including the recA gene [70][65]. The proteins associated with the SOS response play a crucial role in regulating various metabolic processes within bacterial cells [71][66]. Furthermore, mutagenesis is initiated during the advanced phases of the SOS response. The PolV polymerase has been identified as a key driver of SOS-dependent mutagenesis in Escherichia coli (E. coli) [72][67]. The polymerases known for their high error rates in DNA synthesis, which are notorious for their low accuracy, encompass PolV polymerase within their category. One of the causes of induced mutagenesis can be attributed to the activity of PolV, which leads to the insertion of an erroneous nucleotide into the DNA molecule. The formation of a RecA by PolV polymerase is expected to impose certain constraints on the potential diversity of recombinases during the process of selection [73][68]. The process of replicating damaged DNA, known as translesion synthesis (TLS), entails the utilization of PolV polymerase to bypass DNA lesions.
While pol II, pol IV, and polV polymerase are involved in TLS, they also exert an inhibitory effect on RecA-dependent recombination. Achieving equilibrium between these two strategies is of utmost importance. The TLS phenomenon accounts for approximately 1% to 2% of the occurrences in the absence of the SOS response. According to the TLS mechanism, it has been observed that when subjected to stress, TLS has the potential to increase by up to 40% [74][69]. If recombination is performed by specific RecA variants that are efficient in polymerizing onto single-stranded DNA but somewhat impaired in strand invasion, the ratio may also significantly shift in favor of the TLS mechanism [75][70]. Simultaneously, bacteria experience significant detrimental effects due to the rise in mutations. As recombination decreases, there is a subsequent decrease in the size of the bacterial population [75][70]. Moreover, there exists a possibility that moderately unfavorable mutations may undergo fixation when the magnitude of the bacterial population is significantly diminished due to stochastic genetic drift. DNA recombination plays a crucial role in impeding detrimental mutations, as it establishes the boundaries that prevent the occurrence of a “mutational catastrophe” [76][71].
There exist alternative mechanisms for induced mutagenesis, notwithstanding the fact that PolV polymerase (UmuD2C) is the conventional origin of mutations for the purpose of evolutionary selection. While the RecA protein does not engage in interactions to generate a mutasome, an additional error-prone E. coli Pol IV polymerase is synthesized during the SOS response [77][72]. Both polymerases are widely distributed among the majority of bacterial species and belong to the Y family [78][73]. In spite of the substantial diversity observed within the Y family, it is noteworthy that the majority of polymerases share a conserved sequence of 30 residues at their C-terminus. Induced mutagenesis has been observed to occur through the activity of closely related families of polymerases in various bacterial taxa. DnaE2 polymerase, classified as a member of the C family of polymerases, plays a crucial role in the emergence of evolutionary resistance in the bacterium Mycobacterium tuberculosis [79][74].
6. Biofilm-Mediated Resistome
The sensitivity of pseudomonas cells cultivated in biofilms is comparatively lower to antimicrobial agents and host immune responses when compared to cells grown in free aqueous suspension [80][75]. When bacteria proliferate within a biofilm, even those lacking protective mutations or innate resistance mechanisms can exhibit a reduced susceptibility to antibiotics [81][76]. When bacteria experience a loss of biofilm protection, there is a rapid restoration of antibiotic sensitivity. This suggests that the resistance to antibiotics mediated by biofilms is not a result of genetic changes or an adaptive mechanism [82][77]. The overarching mechanisms underlying biofilm-mediated resistance involve impeding the penetration of antibiotics, creating a modified microenvironment that hinders the growth of biofilm cells, triggering an adaptive stress response, and promoting the differentiation of persister cells. These processes collectively serve to safeguard bacteria from the detrimental effects of antibiotic treatment [81][76].
P. aeruginosa synthesizes DNA, proteins, and exopolysaccharides, which are utilized for the formation of a biofilm on the surfaces of lung epithelial cells, leading to the development of persistent lung infections [83][78]. The development of P. aeruginosa biofilms is regulated by multiple factors, primarily including quorum-sensing systems, the GacS/GacA and RetS/LadS two-component regulatory systems, exopolysaccharides, and c-di-GMP [84][79]. Bacterial communication, also known as quorum sensing, regulates the expression of genes in response to changes in the number of cells present [85][80]. P. aeruginosa exhibits three prominent quorum-sensing systems, namely LasI-LasR, RhlI-RhlR, and PQS-MvfR, which collectively contribute to the formation of fully developed and specialized biofilms [86][81]. The GacS/GacA system was found to play a beneficial regulatory role in biofilm development, as evidenced by a tenfold reduction in biofilm generation in a GacA-deficient strain of P. aeruginosa (PA14) compared to the wild-type PA14 strain (Figure 2) [87][82]. The acidification of the environment and upregulation of genes controlled by the PhoPQ and PmrAB two-component regulatory systems were observed as a result of the presence of P. aeruginosa’s environmental DNA (eDNA). This led to a notable increase in aminoglycoside resistance, indicating a previously unknown function of eDNA [88][83]. The intracellular molecule known as c-di-GMP serves as a nucleotide second messenger in the process of signal transduction [89][84]. It plays a role in increasing the levels of c-di-GMP within cells, and these levels are associated with the development of biofilms. In contrast, a diminished level of c-di-GMP has been found to be associated with the presence of planktonic cells [90][85].
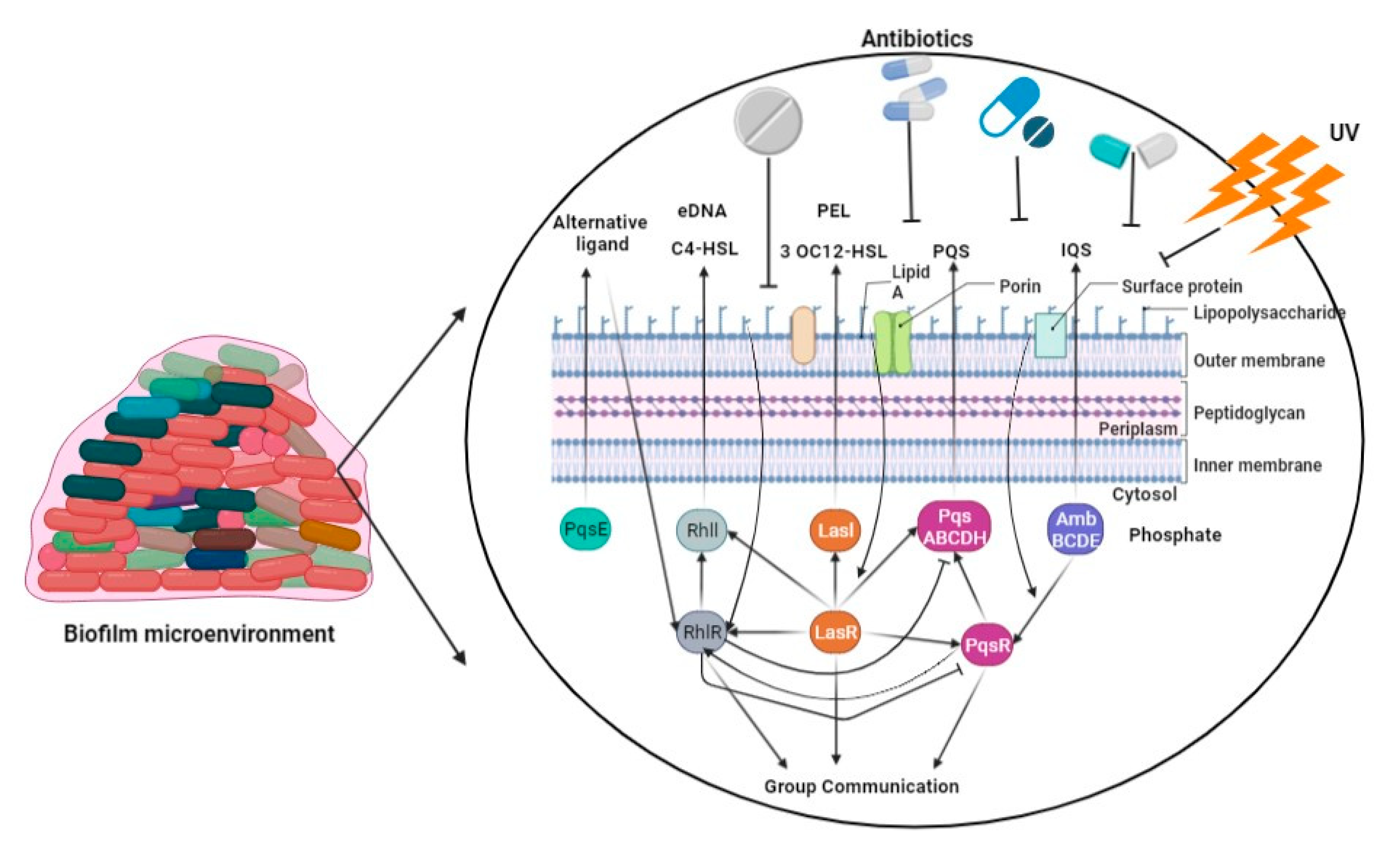
Figure 2. The P. aeruginosa employs four interwoven quorum-sensing loops for biofilm formation using LasI and LasR, RhlI, PqsE and RhlR, PqsABCDH and PqsR, and AmbBCDE.
Throughout the process of biofilm formation, the bacterium P. aeruginosa undergoes numerous changes in its physiological and phenotypic characteristics [91][86]. For example, strains of P. aeruginosa undergo a transformation into a mucoid phenotype during chronic infection in individuals with cystic fibrosis (CF). This transformation is characterized by an enhanced production of alginate, which is stimulated by the specific conditions present in the CF environment. This increased alginate synthesis facilitates the formation of biofilm colonies by the bacteria [92][87]. The periplasmic cyclic β-(1,3)-glucans, with which tobramycin had physical interaction and were sequestered in the periplasm prior to reaching its target site, were produced through the activity of the glucosyltransferase encoded by the ndvB gene [93][88]. An operon encompassing the gene PA14 40260-40230 encodes a novel efflux pump. The resistance of P. aeruginosa to gentamicin and ciprofloxacin in biofilm was observed to decrease upon deletion of the specific operon [94][89]. The regulation of Type VI secretion in P. aeruginosa is governed by the tssC1 gene, which exhibits a high level of expression within biofilm structures [95][90].
References
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426.
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21–22, 41–59.
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327.
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610.
- Skiada, A.; Markogiannakis, A.; Plachouras, D.; Daikos, G.L. Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2011, 37, 187–193.
- Muller, C.; Plésiat, P.; Jeannot, K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 55, 1211–1221.
- Juan, C.; Torrens, G.; González-Nicolau, M.; Oliver, A. Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol. Rev. 2017, 41, 781–815.
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19.
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical Characterization of the Naturally Occurring Oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048.
- Fajardo, A.; Hernando-Amado, S.; Oliver, A.; Ball, G.; Filloux, A.; Martinez, J.L. Characterization of a novel Zn2+-dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2972–2978.
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418.
- Cabot, G.; Ocampo-Sosa, A.A.; Tubau, F.; Macia, M.D.; Rodríguez, C.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; Martínez-Martínez, L.; et al. Overexpression of AmpC and Efflux Pumps in Pseudomonas aeruginosa Isolates from Bloodstream Infections: Prevalence and Impact on Resistance in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 2011, 55, 1906–1911.
- Moya, B.; Dötsch, A.; Juan, C.; Blázquez, J.; Zamorano, L.; Haussler, S.; Oliver, A. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009, 5, e1000353.
- Cabot, G.; Bruchmann, S.; Mulet, X.; Zamorano, L.; Moyà, B.; Juan, C.; Haussler, S.; Oliver, A. Pseudomonas aeruginosa Ceftolozane-Tazobactam Resistance Development Requires Multiple Mutations Leading to Overexpression and Structural Modification of AmpC. Antimicrob. Agents Chemother. 2014, 58, 3091–3099.
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Martín-Pena, M.L.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663.
- Lahiri, S.D.; Alm, R.A. Identification of Novel VEB β-Lactamase Enzymes and Their Impact on Avibactam Inhibition. Antimicrob. Agents Chemother. 2016, 60, 3183–3186.
- Haidar, G.; Philips, N.J.; Shields, R.K.; Snyder, D.; Cheng, S.; Potoski, B.A.; Doi, Y.; Hao, B.; Press, E.G.; Cooper, V.S.; et al. Ceftolozane-Tazobactam for the Treatment of Multi-drug-Resistant Pseudomonas aeruginosa Infections: Clinical Effectiveness and Evolution of Resistance. Clin. Infect. Dis. 2017, 65, 110–120.
- Berrazeg, M.; Jeannot, K.; Enguéné, V.Y.N.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-Lactamase AmpC Increase Resistance of Pseudomonas aeruginosa Isolates to Antipseudomonal Cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255.
- Caballero, J.D.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.W.; Waters, V.J.; Hwang, D.M.; et al. Selective Sweeps and Parallel Pathoadaptation Drive Pseudomonas aeruginosa Evolution in the Cystic Fibrosis Lung. mBio 2015, 6, e00981-15.
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S.; et al. Evolution of the Pseudomonas aeru-ginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 2017, 7, 5555.
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; et al. Deciphering the Resistome of the Widespread Pseudomonas aeruginosa Sequence Type 175 International High-Risk Clone through Whole-Genome Sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423.
- Del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S.; et al. Genomics and Susceptibility Profiles of Extensively Drug-Resistant Pseudomonas aeruginosa Isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17.
- Cabot, G.; Zamorano, L.; Moyà, B.; Juan, C.; Navas, A.; Blázquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa Antimicrobial Resistance and Fitness under Low and High Mutation Rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778.
- Cabot, G.; Florit-Mendoza, L.; Sánchez-Diener, I.; Zamorano, L.; Oliver, A. Deciphering β-lactamase-independent β-lactam resistance evolution trajectories in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 3322–3331.
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of sidero-phore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007.
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778.
- Hocquet, D.; Berthelot, P.; Roussel-Delvallez, M.; Favre, R.; Jeannot, K.; Bajolet, O.; Marty, N.; Grattard, F.; Mariani-Kurkdjian, P.; Bingen, E.; et al. Pseudomonas aeruginosa may accumulate drug resistance mechanisms without losing its ability to cause bloodstream infections. Antimicrob. Agents Chemother. 2007, 51, 3531–3536.
- Solé, M.; Fabrega, A.; Cobos-Trigueros, N.; Zamorano, L.; Ferrer-Navarro, M.; Ballesté-Delpierre, C.; Reustle, A.; Castro, P.; Nicolás, J.M.; Oliver, A.; et al. In vivo evolution of resistance of Pseudomonas aeruginosa strains isolated from patients admitted to an intensive care unit: Mechanisms of resistance and antimicrobial exposure. J. Antimicrob. Chemother. 2015, 70, 3004–3013.
- Riera, E.; Cabot, G.; Mulet, X.; García-Castillo, M.; del Campo, R.; Juan, C.; Cantón, R.; Oliver, A. Pseudomonas aeruginosa carbapenem resistance mechanisms in Spain: Impact on the activity of imipenem, meropenem and doripenem. J. Antimicrob. Chemother. 2011, 66, 2022–2027.
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228.
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305.
- Mulet, X.; Moyá, B.; Juan, C.; Macià, M.D.; Pérez, J.L.; Blázquez, J.; Oliver, A. Antagonistic Interactions of Pseudomonas aeruginosa Antibiotic Resistance Mechanisms in Planktonic but Not Biofilm Growth. Antimicrob. Agents Chemother. 2011, 55, 4560–4568.
- Bruchmann, S.; Dötsch, A.; Nouri, B.; Chaberny, I.F.; Häussler, S. Quantitative Contributions of Target Alteration and Decreased Drug Accumulation to Pseudomonas aeruginosa Fluoroquinolone Resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368.
- Feng, Y.; Jonker, M.J.; Moustakas, I.; Brul, S.; ter Kuile, B.H. Dynamics of Mutations during Development of Resistance by Pseudomonas aeruginosa against Five Antibiotics. Antimicrob. Agents Chemother. 2016, 60, 4229–4236.
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685.
- Greipel, L.; Fischer, S.; Klockgether, J.; Dorda, M.; Mielke, S.; Wiehlmann, L.; Cramer, N.; Tümmler, B. Molecular Epidemiology of Mutations in Antimicrobial Resistance Loci of Pseudomonas aeruginosa Isolates from Airways of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2016, 60, 6726–6734.
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643.
- Gutu, A.D.; Sgambati, N.; Strasbourger, P.; Brannon, M.K.; Jacobs, M.A.; Haugen, E.; Kaul, R.K.; Johansen, H.K.; Høiby, N.; Moskowitz, S.M. Polymyxin Resistance of Pseudomonas aeruginosa phoQ Mutants Is Dependent on Additional Two-Component Regulatory Systems. Antimicrob. Agents Chemother. 2013, 57, 2204–2215.
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crécy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005, 3, e176.
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651.
- Panda, A.; Drancourt, M.; Tuller, T.; Pontarotti, P. Genome-wide analysis of horizontally acquired genes in the genus Mycobacterium. Sci. Rep. 2018, 8, 14817.
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.P. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92.
- Kothari, A.; Kumar, P.; Gaurav, A.; Kaushal, K.; Pandey, A.; Yadav, S.R.M.; Jain, N.; Omar, B.J. Association of antibiotics and heavy metal arsenic to horizontal gene transfer from multidrug-resistant clinical strains to antibiotic-sensitive environmental strains. J. Hazard Mater. 2023, 443, 130260.
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132.
- Patel, G.; Bonomo, R.A. Status report on carbapenemases: Challenges and prospects. Expert Rev. Anti-Infect. Ther. 2011, 9, 555–570.
- Juan, C.; Conejo, M.C.; Tormo, N.; Gimeno, C.; Pascual, Á.; Oliver, A. Challenges for accurate susceptibility testing, detection and interpretation of β-lactam resistance phenotypes in Pseudomonas aeruginosa: Results from a Spanish multicentre study. J. Anti-Microb. Chemother. 2013, 68, 619–630.
- Botelho, J.; Grosso, F.; Quinteira, S.; Mabrouk, A.; Peixe, L. The complete nucleotide sequence of an IncP-2 megaplasmid unveils a mosaic architecture comprising a putative novel blaVIM-2-harbouring transposon in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2017, 72, 2225–2229.
- Van der Zee, A.; Kraak, W.B.; Burggraaf, A.; Goessens, W.H.F.; Pirovano, W.; Ossewaarde, J.M.; Tommassen, J. Spread of Carbapenem Resistance by Transposition and Conjugation Among Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 2057.
- Botelho, J.; Grosso, F.; Peixe, L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J. Antimicrob. Chemother. 2018, 73, 77–83.
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585.
- Del Barrio-Tofiño, E.; Zamorano, L.; Cortes-Lara, S.; López-Causapé, C.; Sánchez-Diener, I.; Cabot, G.; Bou, G.; Martínez-Martínez, L.; Oliver, A.; Galán, F.; et al. Spanish nationwide survey on Pseudomonas aeruginosa antimicrobial resistance mechanisms and epidemiology. J. Antimicrob. Chemother. 2019, 74, 1825–1835.
- Chávez-Jacobo, V.M.; Hernández-Ramírez, K.C.; Romo-Rodríguez, P.; Pérez-Gallardo, R.V.; Campos-García, J.; Gutiérrez-Corona, J.F.; García-Merinos, J.P.; Meza-Carmen, V.; Silva-Sánchez, J.; Ramírez-Díaz, M.I. CrpP Is a Novel Ciprofloxacin-Modifying Enzyme Encoded by the Pseudomonas aeruginosa pUM505 Plasmid. Antimicrob. Agents Chemother. 2018, 62, e02629-17.
- Moya, B.; Zamorano, L.; Juan, C.; Pérez, J.L.; Ge, Y.; Oliver, A. Activity of a new cephalosporin, CXA-101 (FR264205), against be-ta-lactam-resistant Pseudomonas aeruginosa mutants selected in vitro and after antipseudomonal treatment of intensive care unit patients. Antimicrob. Agents Chemother. 2010, 54, 1213–1217.
- Torrens, G.; Cabot, G.; Ocampo-Sosa, A.A.; Conejo, M.C.; Zamorano, L.; Navarro, F.; Pascual, Á.; Martínez-Martínez, L.; Oliver, A. Activity of Ceftazidime-Avibactam against Clinical and Isogenic Laboratory Pseudomonas aeruginosa Isolates Expressing Combinations of Most Relevant β-Lactam Resistance Mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6407–6410.
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62, e01379-18.
- Recio, R.; Villa, J.; Viedma, E.; Orellana, M.; Lora-Tamayo, J.; Chaves, F. Bacteraemia due to extensively drug-resistant Pseudomonas aeruginosa sequence type 235 high-risk clone: Facing the perfect storm. Int. J. Antimicrob. Agents 2018, 52, 172–179.
- Díaz-Cañestro, M.; Periañez, L.; Mulet, X.; Martin-Pena, M.L.; Fraile-Ribot, P.A.; Ayestarán, I.; Colomar, A.; Nuñez, B.; Maciá, M.; Novo, A.; et al. Ceftolozane/tazobactam for the treatment of multidrug resistant Pseudomonas aeruginosa: Experience from the Balearic Islands. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2191–2200.
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In Vivo Emergence of Resistance to Novel Cephalosporin–β-Lactamase Inhibitor Combinations through the Duplication of Amino Acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01117-17.
- Watson, M.E.; Burns, J.L.; Smith, A.L. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology 2004, 150, 2947–2958.
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science 2000, 288, 1251–1253.
- Hocquet, D.; Llanes, C.; Thouverez, M.; Kulasekara, H.D.; Bertrand, X.; Plésiat, P.; Mazel, D.; Miller, S.I. Evidence for induction of integron-based an-tibiotic resistance by the SOS response in a clinical setting. PLoS Pathog. 2012, 8, e1002778.
- Breidenstein, E.B.M.; Bains, M.; Hancock, R.E.W. Involvement of the Lon Protease in the SOS Response Triggered by Ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2012, 56, 2879–2887.
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 2020, 19, 331–342.
- Smith, P.A.; Romesberg, F.E. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat. Chem. Biol. 2007, 3, 549–556.
- Horii, T.; Ogawa, T.; Nakatani, T.; Hase, T.; Matsubara, H.; Ogawa, H. Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 1981, 27, 515–522.
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64.
- Sutton, M.D.; Smith, B.T.; Godoy, V.G.; Walker, G.C. The SOS response: Recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 2000, 34, 479–497.
- Gruber, A.J.; Erdem, A.L.; Sabat, G.; Karata, K.; Jaszczur, M.M.; Vo, D.D.; Olsen, T.M.; Woodgate, R.; Goodman, M.F.; Cox, M.M. A RecA Protein Surface Required for Activation of DNA Polymerase V. PLoS Genet. 2015, 11, e1005066.
- Naiman, K.; Pagès, V.; Fuchs, R.P. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res. 2016, 44, 7691–7699.
- Yakimov, A.; Bakhlanova, I.; Baitin, D. Targeting evolution of antibiotic resistance by SOS response inhibition. Comput. Struct. Biotechnol. J. 2021, 19, 777–783.
- Sniegowski, P.D.; Gerrish, P.J.; Lenski, R.E. Evolution of high mutation rates in experimental populations of E. coli. Nature 1997, 387, 703–705.
- Pomerantz, R.T.; Kurth, I.; Goodman, M.F.; O’Donnell, M.E. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat. Struct. Mol. Biol. 2013, 20, 748–755.
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8.
- Boshoff, H.I.M.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193.
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138.
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113.
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323.
- Taylor, P.K.; Yeung, A.T.; Hancock, R.E. Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies. J. Biotechnol. 2014, 191, 121–130.
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The Formation of Biofilms by Pseudomonas aeruginosa: A Review of the Natural and Synthetic Compounds Interfering with Control Mechanisms. Biomed Res. Int. 2015, 2015, 759348.
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199.
- Kang, D.; Turner, K.E.; Kirienko, N.V. PqsA Promotes Pyoverdine Production via Biofilm Formation. Pathogens 2017, 7, 3.
- Parkins, M.D.; Ceri, H.; Storey, D.G. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm for-mation. Mol. Microbiol. 2001, 40, 1215–1226.
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 544–553.
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273.
- Ha, D.-G.; O’Toole, G.A.; Ghannoum, M.; Parsek, M.; Whiteley, M.; Mukherjee, P.K. c-di-GMP and its Effects on Biofilm Formation and Dispersion: A Pseudomonas aeruginosa Review. Microbiol. Spectr. 2015, 3, MB-0003-2014.
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219.
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid Pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34.
- Mah, T.-F.; Pitts, B.; Pellock, B.; Walker, G.C.; Stewart, P.S.; O’Toole, G.A. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 2003, 426, 306–310.
- Zhang, L.; Mah, T.-F. Involvement of a Novel Efflux System in Biofilm-Specific Resistance to Antibiotics. J. Bacteriol. 2008, 190, 4447–4452.
- Zhang, L.; Hinz, A.J.; Nadeau, J.P.; Mah, T.F. Pseudomonas aeruginosa tssC1 links type VI secretion and biofilm-specific antibiotic resistance. J. Bacteriol. 2011, 193, 5510–5513.
More