Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Thabiso Victor Miya and Version 2 by Camila Xu.
Prostate cancer (PCa) continues to be the most diagnosed cancer and the second primary cause of fatalities in men globally. There is an abundance of scientific evidence suggesting that the human microbiome, together with its metabolites, plays a crucial role in carcinogenesis and has a significant impact on the efficacy of anticancer interventions in solid and hematological cancers.
- prostate cancer
- human microbiome
- microbiota
- short-chain fatty acids
1. Introduction
Prostate cancer (PCa) is the second most diagnosed cancer among men worldwide, after lung cancer [1]. In addition, PCa is also the second most common cause of cancer-induced fatalities, globally. In 2020, PCa was estimated to account for a total of 1,414,259 new cases and 375,304 related fatalities worldwide [2]. Although there is currently no cure for advanced PCa, the five-year survival rate of localized PCa is >99%, [3] [4]. Advanced PCa is defined by recurrence post definitive local therapy such as radiation and/or surgery, or with evidence of metastases at any point [5]. Conversely, metastatic PCa has a 31% five-year survival rate [4] and thus, there is an imperative need for effective novel therapeutic agents against this non-curable disease [5]. However, management of PCa is rapidly changing due to advancements in understanding of its evolution, mutational landscape, and signaling pathways, as well as its resistance mechanisms. In recent years, intense research has focused on the indirect or direct link between cancer and particular microflora of various cancers, PCa included [6].
2. The “Prostate Microbiome”
2.1. Intraprostatic Microbiome
Several research studies have identified microbial composition in PCa tissues. However, it is still unclear if the “prostate microbiome” is unique [7][8][27,28]. The challenge with these studies is contamination that ends up giving false positive results [9][29]. Studies that analyzed the presence of “prostate microbiome” have thus far reported that the prostate microflora composition is the same as in the urethra [10][11][30,31]. Hochreister and colleagues carried out 16S rRNA PCR to ascertain the presence or absence of bacterial species in prostate tissues collected from 9 patients and 18 healthy controls. The study detected a significant presence of bacterial deoxyribonucleic acid (DNA) in PCa samples compared to healthy controls. However, the researchers did not identify which bacterial species the detected DNA belonged to [12][32]. Sfanos et al. [13][33] caried out 16S ribosomal DNA (rDNA) sequencing on radical prostatectomy tissue core specimens. Notably, bacterial DNA was observed in the prostate tissues. However, when the results were contrasted to the core samples, the specimens were negative. Additionally, there was no significant link observed between bacterial species presence and chronic or acute inflammation. Interestingly, focal regions showed numerous bacterial species that are usually found in urinary-tract infections, these included Pseudomonas, Enterococci, and Escherichia spp. Furthermore, the authors observed other species such as Actinomyces, Acinetobacter, and Streptococcus spp. which are frequently present in the urethral flora in physiological settings. Based on these findings, the authors concluded that the prostate microbiome might not exist and that their findings were merely due to remnant bacterial DNA which was ‘fossilized’ in the prostate [13][33]. In 2017, another study was conducted in which 16S rDNA next-generation sequencing (NGS) and RNA sequencing were carried out on specimens from 20 patients with an aggressive PCa [7][27]. The aim of this study was to determine histopathologically the existence of infectious agents in aggressive PCa cases. The researchers reported the existence of the Enterobacteriaceae family, of which Propionibacterium acnes (P. acnes) and Escherichia were the most predominant species [7][27]. All these studies have paved the way for identification of the microbiomes and research into their potential role during PCa pathogenesis [9][29]. Cavarretta et al. [10][30] observed that P. acnes was the most common bacterial species in PCa patients, and that it was evenly present in all tissues. The role of P. acnes in the pro-inflammatory pathway within prostate tissue was confirmed using murine models. These results suggest that P. acnes may play a central role in PCa carcinogenesis [14][15][34,35]. Additionally, the authors also observed a larger amount of Staphylococcaceae in malignant tissues and also a larger proportion of Streptococcaceae in non-malignant tissues [10][30]. It has been speculated that the existence of Streptococcus in non-malignant tissues might be a sign of normal microbiome of a healthy prostate tissue [9][29]. However, it is critically important to note that Staphylococcus and Streptococcus spp. are among the most common human skin bacteria. Therefore, they represent contamination during laboratory analysis [16][36]. Feng and colleagues carried out the analysis of tissue specimens from a cohort of 65 patients who underwent radical prostatectomy [8][28]. In this study, Escherichia, Cutibacterium, Pseudomonas, and Acinetobacter were reported to be the most predominant bacterial species in the examined tissues. In addition, the authors did not observe any difference in the adjacent benign tissues [8][28]. In another study, the presence of pathogens in tissue specimens from a cohort comprising 50 patients who underwent radical prostatectomy and a cohort of 15 benign prostatic hyperplasia (BPH) patients who had undergone prostate transurethral resection procedure were evaluated [17][37]. This was conducted using pan-pathogen microarray metagenomics analysis (PathoChip). The authors reported a distinct pathogenic microbiome in the specimens from PCa patients. This pathogenic microbiome comprised Firmicutes, Bacteroides, Actinobacteria, and Proteobacteria phyla. Furthermore, there was no difference between the microbiota signatures of PCa patients’ samples and samples from those without PCa. Nonetheless, the most important observation was the discovery of Helicobacter pylori in >90% of PCa samples, which further confirmed the integration of H. pylori-cytotoxin-associated gene A (CagA) into the DNA of the prostate tumor [17][37]. The CagA gene has been reported as a virulence factor for H. pylori and has been linked to gastric cancer pathogenesis via suppression of tumor suppressor genes (TSGs) and actuating of proto-oncogenes [18][38]. The authors also reported the existence of numerous oncogenic viruses such as human papilloma virus (HPV) 16, HPV 18, and human cytomegalovirus which accounted for 41% of all isolated viruses [17][37]. In the same year, Miyake et al. [19][39] assessed the existence of pathogens involved in sexually transmitted infections (STIs) and their role in PCa carcinogenesis. The authors collected samples from 45 PCa and 33 BPH patients and analyzed them for the presence of numerous pathogens. The analyzed pathogens included Ureaplasma urealyticum, Mycoplasma genitalium, Chlamydia trachomatis, Mycoplasma hyorhinis, Neisseria gonorrhoeae, and HPV 18/16. The authors reported that Mycoplasma genitalium was the only species linked with a higher Gleason score and PCa pathogenesis [19][39]. In 2009, a meta-analysis involving case-control research studies from patients with PCa and healthy controls (HCs) was carried out [20][40]. The authors reported a significant link between cancer risk and infection history of STI such as HPV and Mycoplasma genitalium [20][40].2.2. Genitourinary Microbiome
For a long time, the urinary tract was thought of as a sterile organ [21][22]. However, numerous research studies have recently reported the existence of a urinary microbiome that is different from the gut microbiome [22][23][24][24,41,42]. This urinary-tract microbiome signifies the effect of the microbiome in PCa [21][22]. Since the urinary tract is in close proximity to the prostate gland, it can contaminate it and thus, urinary microbial research studies are crucial in the identification of prostate diseases [25][26][7,43]. Numerous studies have isolated different microbial strains from the urine of adult males. These microbes include Staphylococcus, Prevotella, Finegoldia, Streptococcus, Veillonella, Propionibacterium, and Corynebacterium [27][28][29][44,45,46]. P. acnes can be described as a proinflammatory bacterium that is commonly isolated from the urine of males. The link between P. acnes, human PCa, and prostatitis in animal models has previously been reported [13][14][30][31][32][33][34][26,33,34,47,48,49,50]. Chronic prostatitis is commonly induced by uropathogenic strains of Enterococci and E. coli [35][51]. On the other hand, prostatitis induced by P. acne and E. coli strains can result in hyperplasia and morphological changes [35][51]. Furthermore, these changes have also been implicated in decreasing a tumor suppressor called NKX 3.1 in the prostate [14][34]. Two clinical studies have reported that proinflammatory bacteria such as Propionimicrobium lymphophilum, Anaerococcus lactolyticus, Streptococcus anginosus, and Varibaculum cambriense are frequent in patients with cancer [36][37][52,53]. These results suggest that pro-inflammatory bacteria can potentially induce inflammation on the prostate gland for PCa pathogenesis [38][54]. Several studies have reported that the human urinary microbiome differs according to gender, disease, and age. However, these studies were different with regard to methodology and sample collection method, as well as inclusion criteria [22][24][29][24,42,46]. For instance, it was previously reported that the genera Streptococcus, Staphylococcus, and Corynebacterium are mainly found in the male urinary microbiome [22][30][24,26]. Dysbiosis of the urinary microbiome can be due to factors such as urinary incontinence, puberty, or antimicrobial agents of prostatic secretions, as well as sexual behavior [39][40][55,56]. Dysbiosis plays a central role in terms of reaction to urinary-tract diseases, and also affects immune molecules [41][42][57,58]. In addition, urinary microbiome variation between individuals plays a significant role in the susceptibility to STIs such as N. gonorrhoeae and C. trachomatis [27][44]. Clinical studies have also shown increased levels of PSA associated with STIs, which could indicate involvement of the prostate [43][44][59,60]. Additionally, the PCa pathogenesis risk can be exacerbated by a history of inflammatory STIs [45][61]. Association between male urinary microbiome and prostate diseases such as PCa, BPH, and prostatitis has been reported, and has been discussed elsewhere [11][30][46][26,31,62]. However, more clinical studies that focus on the effects of the urinary microbiome composition on PCa carcinogenesis need to be carried out [47][63].2.3. Gut Microbiome
Numerous studies have established a potential link between gut microbiome and PCa development and resistance to anticancer therapies [48][64]. Liss et al. [49][65] analyzed gut microbiome from 133 USA men who had undergone prostate biopsy. Analysis were carried out on rectal swabs using 16S rRNA sequencing. The authors reported that Streptococcus and Bacteroides spp. were higher in PCa patients. They then carried out metagenome analysis which showed that the gut-microbiome arginine and folate pathways were significantly modified. Subsequently, the authors suggested that the risk of PCa may potentially be impacted by the gut bacteria [49][65]. Golombos [50][66] analyzed the gut microbiota from a cohort of 20 men. Of the 20 men, 12 had high-risk PCa, while 8 had benign prostate hypertrophy [50][66]. Results demonstrated that the men with PCa had elevated Bacteroides massiliensis. Nevertheless, the specific mechanism of action is not yet fully understood [50][66]. Matsushita and colleagues analyzed the gut microbiome from a cohort of 152 Japanese men who had undergone prostate biopsy [51][67]. The analysis showed that Alistipes, Rikenellaceae, and Lachnospira were highly elevated in men with increased Gleason PCa. The gut microbiota profile, consisting of 18 gut bacteria, could be used to predict PCa with a higher Gleason score compared to the prostate-specific antigen (PSA) test. In this study, bacterial taxa within high-risk PCa were not impacted by metastasis. These observations suggest that modifications in the gut microbiome in high-risk PCa induces PCa and are not the result of PCa [51][67].3. Microbiome-Derived Short-Chain Fatty Acids (SCFAs)
It is widely known that cancer is induced by the interaction between host genetics and environmental factors. However, several research studies have highlighted the crucial role that microorganisms play in carcinogenesis [52][68]. Many carcinogenic microbes such as Helicobacter pylori, hepatitis B (HBV) and C (HCV), and HPV have been identified in 20% of all malignancies [53][69]. An additional group of oncogenic microbes, called the microbiota, were recently reported as key factors in carcinogenesis [52][68]. Recent years have seen the development of cutting-edge technologies to analyze and quantify human microbiota and link their role in carcinogenesis [54][70]. Despite these ongoing investigations, the exact role of the microbiota in carcinogenesis is yet to be fully understood [55][71]. However, studies have shown that bacterial-derived metabolites are the central link between gut microbiota and cancer development [56][72]. Gut microbiota converts fermentable and non-digestible carbohydrates such as dietary fiber into several SCFAs (Figure 12) [57][73]. Predominant SCFA members are acetate, butyrate, and propionate (Figure 12). The total intestinal concentration of these SCFAs may reach over 100 mM [58][74].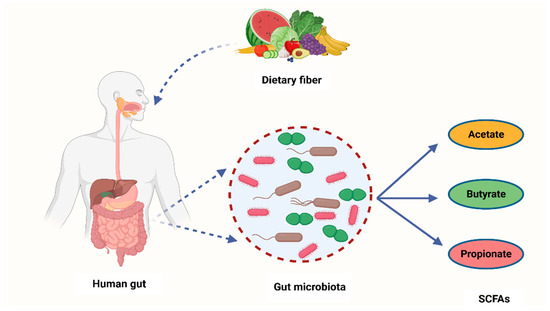
Figure 12. A schematic diagram showing how bacteria from the human microbiome convert dietary fiber into three main SCFAs—acetate, butyrate, and propionate. Acetate is synthesized through conversion of pyruvate. This occurs in three ways: either through the reductive acetyl-CoA pathway, the Wood–Ljungdahl pathway, or directly through acetyl-CoA. On the other hand, butyrate is a product of acetyl-CoA reduction to butyryl CoA, which subsequently becomes converted into butyrate-by-butyrate kinase and transbutyrylase enzymes. Moreover, butyryl CoA may form butyrate through butyryl-CoA transferase-acetate. Lastly, propionate is formed through the succinate pathway. It can also be synthesized via the acrylate pathway from lactate (precursor) with hexoses and pentoses (simple sugars) acting as reaction substrates. Propionate can also be formed through the propanediol pathway whereby rhamnose and fucose (deoxyhexoses) function as substrates. SCFAs—short-chain fatty acids, Acetyl-CoA—acetyl coenzyme A. Created with BioRender.com (Accessed on 4 July 2023).
3.1. The Mechanism of SCFAs in Cells
SCFAs display intracellular and extracellular outcomes via binding ligands to their receptors and also functioning as epigenetics modulators. SCFA receptors are found throughout the human body, and they belong to G protein-coupled receptors (GPCRs). This suggests their role in numerous cellular pathways [71][87]. For example, a surface receptor of macrophages, colonocytes, and adipocytes called GPR109A is frequently involved in the release of fat deposits in deprivation conditions in adipocytes [72][73][88,89]. The reduction of GPR109A expression can result in CRC pathogenesis. GPR109A receptor has been reported to be involved in T-reg-cell differentiation and establishment of proinflammatory (IL-18) and anti-inflammatory (IL-10) cytokines [72][88]. These responses were previously linked with cancerous outcomes, as demonstrated in Niacr1-/- mice [74][90]. In addition, SCFAs can also affect apoptosis and cell-cycle modulation [56][72]. On the other hand, activation of GPR41/43 receptor in MCF-7 cells promotes Ca2+ intracellular levels and stimulation of MAPK p38 [56][72]. These observations are widely associated with carcinogenesis and cell stress responses [75][76][91,92]. Furthermore, GPR43 receptor is absent in colon tumors and metastatic cells which suggests its role in oncogenesis. Apoptosis and G0/G1 cell cycle arrest were observed after the restoration of GPR43 expression in adenocarcinoma cell lines [77][93]. A potential link between SCFAs and cancer development may be due to GPCR stimulation which can subsequently activate cascades of responses resulting in cancer or prevention thereof [56][72]. SCFAs can also function as ligands to receptors located in the membrane, thus impacting cell metabolism. Butyrate may use sodium-coupled monocarboxylate transporter (SMCT1) during entry into the host cell. SMCT1 was initially classified as a possible tumor suppressor [56][78][72,94]. Butyrate can also utilize other carriers to propagate into the body [56][72]. Monocarboxylate transporter 4 (MCT4) is one of the transporters which fuses butyrate to the blood flow [79][95]. This enables butyrate to showcase a systemic effect on the host organism inside the bloodstream. Moreover, butyrate is able to enter back into the intestinal cavity using breast cancer resistance protein (BCRP) [56][72]. Decrease in BCRP mRNA expression has a potential association with colorectal adenoma pathogenesis which can possibly be linked with butyrate build-up inside the cells [80][96]. Post cell entry, butyrate can have an impact on histone deacetylases (HDACs) [56][72]. HDACs are mainly involved in cell-cycle modulation, cell proliferation, and apoptosis [81][97]. Furthermore, butyrate, together with its binding to HDACs, has been linked to CRC pathogenesis [81][82][97,98]. HDAC enzymatic activity becomes inhibited once butyrate binds to it. This results in gene expression modification and histone hyperacetylation. Butyrate can suppress tumorigenic cell growth via cell-cycle arrest activation and programmed cell death [56][72]. Lastly, butyrate might have an impact on other processes involved in epigenetic modulation, including DNA methylation, hyperacetylation of non-histone proteins, and histone methylation and phosphorylation [56][83][84][72,99,100].3.2. The Role of SCFAs in PCa Carcinogenesis
The gut-microbiota-derived SCFAs contribute to the modulation of HDACs [85][101]. In return, this SCFA-mediated HDAC modulation may play a crucial role in cell homeostasis. This is because HDACs have an impact on immune-cell migration, cell adhesion, programmed cell death, chemotaxis, and cytokine synthesis. As such, manipulation of intestinal-tract SCFA levels through the alteration of the microbiota can be a potential strategy for cancer treatment and prevention. Notably, it was shown that gastric cancer and breast cancer are mediated in individuals with decreased proportions of SCFAs in feces or individuals on a diet low with SCFAs [66][82]. SCFAs can block cell growth, migration, HDACs, and activate apoptosis. In turn, these SCFA properties enable it to reduce cancer incidence [55][71]. Prior to being recognized as a microbiota-derived metabolites, SCFAs were studied as differentiation or an antiproliferative agents for treating solid tumors; for example, breast cancer and PCa [86][102]. Samid et al. [87][103] were the first researchers to study the impact of acetate on PCa. In this study, dose-dependent cell proliferation inhibition was observed after DU145 (hormone-refractory PCa cell line), LnCap (a HSPC cell line), and PC3 cell lines were exposed to phenylacetate (PA) [87][103]. Furthermore, tumors were not observed after PC3 cells were treated with PA and transplanted into nude mice. These results demonstrated antitumor capabilities of acetate [87][103]. On the other hand, Carducci et al. [88][104] studied the impact of butyrate on PCa development. The authors observed that the apoptotic and growth-inhibitory effects of butyrate were much higher compared to those of acetate in PCa cell lines [88][104]. Since these clinical studies involved a low number of participants, they were not enough to draw solid conclusions regarding acetate and butyrate effectiveness against PCa development [89][90][105,106]. Conversely, research studies have recently reported the association between SCFAs and PCa progression [85][101]. For example, Matsushita et al. [91][107] reported that SCFAs promoted PCa growth through IGF1 signaling in Pten knockout mice [91][107]. In this study, Pten knockout mice were utilized as a PCa model to investigate the link between animal-fat consumption and PCa pathogenesis as mediated by gut microbiota. This is because animal fat and subsequent obesity are the crucial risk factors for PCa pathogenesis, and gut microbiota composition varies with dietary composition as well as body type. Antibiotic mixture (Abx) was orally administered in these PCa mice. In addition, the mice were fed a high-fat diet (HFD) which contained high lard quantities. This resulted in a significant alteration of the gut microbiome composition, including Clostridiales and Rikenellaceae species and blocked PCa cell proliferation. It also reduced levels of circulating IGF1 and the expression of prostate Igf1 gene. On the other hand, Abx exposure suppressed phosphatidylinositol-3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) activities downstream of the IGF1 receptor in the prostate tissue. In the same study, proliferation of 22Rv1 and DU145 PCa cell lines was directly promoted by IGF1. Abx exposure also decreased levels of fecal SCFAs produced by the intestinal bacteria. On the other hand, SCFA supplementation promoted tumor growth through the increasing of IGF1 levels. Notably, IGF1 was reported to be highly expressed in PCa tissue from patients with obesity. The authors concluded that IGF1 synthesis, as stimulated by gut-microbiota-derived SCFAs, promotes PCa by activating localized PI3K and MAPK signaling pathways (Figure 23) [91][107].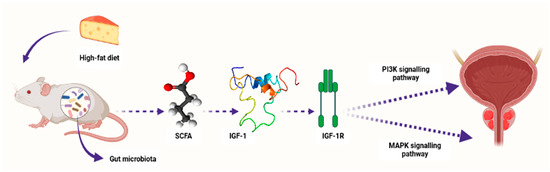
Figure 23. A schematic diagram showing how microbiota-derived SCFAs induced PCa carcinogenesis by activating the PI3K and MAPK signaling pathways through IGF1 and its receptor IGF-1R in Pten knockout PCa mice. Composition of the gut microbiota is influenced by diet content, e.g., high-fat diet. SCFAs—short-chain fatty acids, IGF-1—insulin-like growth factor-1, IGF-1R—insulin-like growth factor-1 receptor, PI3K—phosphatidylinositol-3 kinase, MAPK—mitogen-activated protein kinase. Created with BioRender.com (Accessed on 31 July 2023).
3.3. The Effect of Microbiome-Derived SCFAs on Response to Cancer Immunotherapy
State-of-the-art techniques have shown that microbiota influence carcinogenesis and immunotherapy [93][109]. Moreover, microbiota can positively or negatively impact tumorigenesis [94][110]. Bacteria synthesize cancerous compounds or toxins which can result in inflammatory or immunosuppressive responses that support carcinogenesis [93][109]. Conversely, gut microbiota may inhibit oncogenesis by boosting antitumor immunity [94][110]. In addition, gut microbiota impair anticancer treatment and toxicity efficacy by changing systemic and local immune responses [95][111]. Analysis of microbial-derived metabolites such as SCFAs, as well as gut microbiota imbalance with regard to their effects on immune responses, will ameliorate understanding of numerous frequent etiological disorders [96][112]. SCFAs, particularly propionate, butyrate, and acetate, are found in certain concentrations. Furthermore, SCFA amounts can change with age, disease, and diet. SCFA concentrations are particularly regulated by the gut microbiota proportions. In addition, gut dysbiosis can induce imbalance of the synthesized SCFAs. It was also observed that SCFAs can inhibit HDAC activity which is involved in deacetylation and histone crotonylation. These SCFA attributes can support pro/anti-inflammatory hemostasis and potentiate their immunomodulatory capabilities. SCFAs influence gut immune cells and modulate the immune system through multiprotein inflammasome complexes. They also have localized functions in the intestines which are occupied by gut bacteria [96][112]. Furthermore, SCFAs are important for immune regulation [97][113]. Butyrate has systemic anti-inflammatory properties through alteration of cytokine expression of immune cells and having an impact on cellular processes such as activation, propagation, and apoptosis. Butyrate particularly inhibits HDAC. On the other hand, HDAC prohibits gene transcription by retaining the compact structure of the chromatin. Therefore, inhibition of HDAC by butyrate leads to hyperacetylation. In this way, butyrate can regulate gene expression and exert its antiproliferative properties. (Figure 34) [98][114].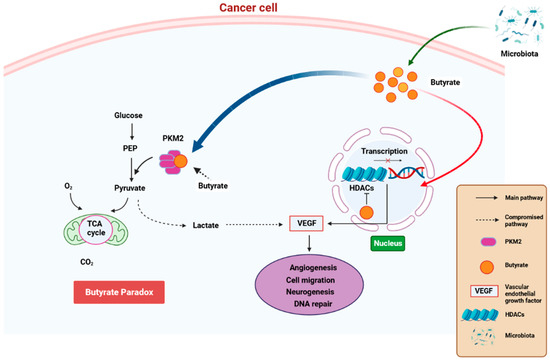
Figure 34. A schematic diagram showing how butyrate exerts anti-cancer properties. Butyrate is synthesized is a by-product of fiber fermentation by microbiota and then accumulates in cancerous gastric epithelia through the Warburg effect. This result in glucose metabolism and increased lactate synthesis by the epithelial cells. In return, lactate induces processes that trigger the upregulation of VEGF, resulting in the upregulation of angiogenesis, cell migration neurogenesis, and aberrant DNA repair. These processes result in cancer pathogenesis. Butyrate effectivity against cancer occurs through the butyrate paradox in two ways: (1) butyrate travels from the cytoplasm to the nucleus where it acts as an HDAC inhibitor by terminating cell-cycle continuation via altered gene expression and (2) butyrate can reverse metabolism anaerobic glycolysis to OXPHOS by binding to PKM2. This, in return, renders PKM2 a more active dephosphorylated tetrameric version, thus favoring energy creation via the Krebs cycle. HDAC—histone deacetylase, OXPHOS—oxidative phosphorylation, PKM2—pyruvate kinase M2. Created with BioRender.com (Accessed on 25 June 2023).