1. Introduction
Organo-fluorinated drugs have been one of the most rapidly growing classes of organic compounds over the last 20 years
[1][2][3][4][5][6][7][8][9][10][11][1,2,3,4,5,6,7,8,9,10,11]. The role of fluorine in the design of pharmaceutical drugs
[12][13][14][15][16][17][18][19][20][21][22][23][24][12,13,14,15,16,17,18,19,20,21,22,23,24], agrochemicals, and specialty materials is widely recognized
[25][26][27][28][29][25,26,27,28,29]. Fluoro-pharmaceuticals
[3] are compounds of diverse nature containing at least one fluorine atom or a fluorinated functional group (e.g., trifluoromethyl, CF
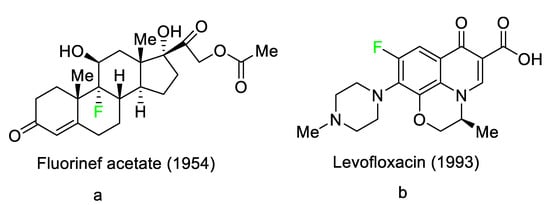
3). These pharmaceuticals have accounted for an estimated 20% of commercialized medications in recent years. Florinef acetate, the first fluoro-pharmaceutical, was introduced into the market in 1954 (
Figure 1a). Florinef acetate is a corticosteroid synthesized with a fluorine atom in the stereogenic 9 position. It has strong mineralocorticoid properties as well as high glucocorticoid activity, and it is used to treat adrenogenital syndrome, adrenal insufficiency, and postural hypotension
[30]. Fluoroquinolones (new quinolones) were released in the 1980s and they are another historically significant group of fluoro-pharmaceuticals (
Figure 1b). Fluoroquinolones are effective antibacterial agents because they inhibit the activity of DNA gyrase and topoisomerase. This mechanism of action is fundamentally different from that of lactam antibiotics such as penicillin, cephalosporin, and antibacterial sulfa drugs. The number of fluoro-pharmaceuticals to receive FDA approval has continuously increased over the last two decades owing to the success of fluorinated corticosteroids and fluoroquinolones. Almost 300 fluoro-pharmaceuticals have been registered worldwide
[30][31][32][30,31,32].
Figure 1.
Selected fluoro-pharmaceuticals: (
a
) Fluorinef acetate, (
b
) Levofloxacin.
Several factors can explain the high occurrence of fluoro-organic molecules in medications. First and foremost, given that the fluorine (F, van der Waals radius of 1.47 Å) atom is slightly larger than the hydrogen (H, van der Waals radius of 1.20 Å) atom, it does not significantly alter the parent structure when H is replaced by F in a drug candidate
[33]. Second, the C–F bond is the strongest link that carbon can make and it often boosts the metabolic stability of fluoro-pharmaceuticals. Third, as the most electronegative element (3.98), F causes bond polarization, which can affect the lipophilicity/hydrophilicity balance of a compound. Finally, F is a moderate hydrogen acceptor and the hydrogen bond-accepting analogy would imply a carbonyl moiety, which is especially pertinent given that the dipoles of C–F and C–O are sometimes comparable. Although the combination of these distinct features of F has a minor impact on the absorption, distribution, metabolism, and excretion of therapeutic candidates, these effects are likely to be complex and require further attention
[34][35][34,35].
According to probability theory, the constant success of fluoro-pharmaceuticals implies that, for medicinal chemists, choosing fluoro-organic compounds is a viable method to considerably reduce the chance of unsuccessful trial-and-error attempts during drug discovery
[36].
Drug discovery is a difficult, high-risk, expensive, and time-consuming process, with a success rate for small compounds estimated at 1/20,000 or 1/30,000
[36]. Although significant progress has been made in this field thanks to computer-aided approaches such as molecular modeling, these methods are still too immature for successful drug design, as evidenced by surprising failures in subsequent clinical phases
[37].
2. FDA-Approved Fluorinated (F-18) Radiolabeled Drugs
The development and progress of positron emission tomography (PET) imaging agents labeled with the radionuclide F-18 for uses spanning from fundamental research to clinical imaging have evolved rapidly. F-18 is the most appealing PET radionuclide for labeling small molecules because F can be easily incorporated without interfering with the biological activity of a compound, and the 110 min physical half-life of F-18 allows sufficient time for labeling, purification, quality control, and regional distribution to PET centers without on-site radiosynthetic capabilities. These
18F-labeled tracers have been utilized to image a variety of metabolic and biochemical processes related to cancer, Alzheimer’s disease, Parkinson’s disease, and cardiovascular illnesses. The most widely used radiopharmaceutical is
18F-fluoro deoxyglucose (FDG), which can indicate glucose absorption and energy use in diverse cells and has applications in oncology and neuroscience. FDG, like glucose, is taken up into cells by glucose transporters and is phosphorylated at the 1-position by hexokinase. However, after rapid phosphorylation, FDG-6-phosphate cannot be metabolized further because the F atom in position 2 prevents this, resulting in it being retained within the cell. Therefore, the intracellular FDG concentration is proportional to the glucose utilization of the tissue
[38][39][38,39]. This is useful in oncology because tumor cells have a faster glycolytic rate and, hence, absorb FDG more actively than healthy cells.
FDG-PET can be used in clinical contexts such as initial diagnosis, assessment of response to therapy, and detection of recurrence. Lung nodules, non-small-cell lung carcinomas, lymphomas, melanomas, colorectal malignancies, and head and neck tumors are the tumor types most commonly studied with FDG-PET
[40]. This technique has also been used in research and clinical practice to quantify cerebral glucose metabolism
[41]. In clinical neurology, PET imaging can be used to perform differential diagnosis, define pathophysiological changes in the course of a disease, monitor illness, and also for follow-up purposes. FDG-PET can detect impaired glucose metabolism in a variety of disorders, including Alzheimer’s disease, Creutzfeldt–Jakob disease, and epilepsy
[42]. Between 2016 and 2022, the FDA approved the following five fluorinated drugs containing F-18.
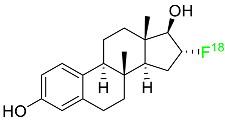
2.1. Flortaucipir F-18
Flortaucipir F-18 is a small lipophilic tracer capable of crossing the blood–brain barrier and binding to aggregated tau proteins. It is used in PET imaging for the diagnosis of Alzheimer’s disease (
Table 1). Flortaucipir F-18 received medical approval for use in the US in May 2012. In the diagnosis of Alzheimer’s disease in adults, this radioactive tracer is used to image aggregated neurofibrillary tangles (NFTs) using PET. Patients who are receiving treatment for chronic traumatic encephalopathy should not use this drug. Flortaucipir F-18 rapidly penetrates the blood–brain barrier, circulates in the body, and binds to NFTs. After binding, the subsequent radioactive decay releases pairs of 511 keV gamma rays that are useful for imaging diagnostics. Alzheimer’s disease is diagnosed using the pattern and intensity of emission during imaging. The kidneys are the primary organs through which flortaucipir F-18 is removed. The original drug and four uncharacterized metabolites were first studied in mice. All metabolites were discovered in the kidneys and liver, except metabolite 2, which was detected only in the liver. Patients who overdose are more likely to experience severe side effects such as headaches and elevated blood pressure. It is advised to take symptomatic and supportive measures. Flortaucipir F-18 has an estimated half-life in plasma of 17.0 +/− 4.2 min
[43].
Table 1.
FDA-Approved Fluorinated (F-18) Radiolabeled Drugs.
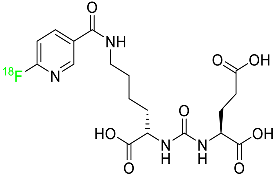
2.2. Fluciclovine F-18
Fluciclovine F-18 is a synthetic amino acid that detects the upregulation of amino acid transport, especially in prostate cancer (Table 1). It is used for the identification of suspected sites of prostate cancer and it provides signals in 3 to 5 min.
Its miniature shape facilitates its uptake by tumor cells through its amino acid transporter without interacting with the body’s metabolic system. An individual suspected of having prostate cancer is expected to show an increased level of blood-specific prostate antigens. The overexpressed L-type amino acid transporters LAT1 and LAT3, which respond to binding with essential amino acids, play a significant role in the tumoral mechanisms of cell proliferation. Fluciclovine F-18 is transported into prostate cancer cells via ASCT1 (Alanine Serine Cysteine Transporter 1) and LAT1 (Large neutral amino acid transporter) injected intravenously. When an environment is acidic, the primary function of LAT1 is boosted. When injected into a human, fluciclovine F-18 takes roughly 2 to 10 min to reach the target cells. Additionally, within 90 min of the injection, a 63% reduction in mean tumor size is achieved. This drug is distributed throughout various organs such as the liver (14%), bone marrow (12%), lungs (7%), myocardium (4%), and pancreas (3%). The major characteristic of fluciclovine F-18 is that it is incapable of binding to plasma proteins and it does not participate in the synthesis of new proteins. Regarding its removal, 3% of the dose is eliminated through urine in the first four hours post-injection and 5% in the next twenty-four hours. One of the primary merits of fluciclovine F-18 is that it has the potential to be mutagenic but not carcinogenic
[44].
2.3. Fluorodopa F-18
A fluorinated analogue of levodopa called fluorodopa F-18 is used in PET diagnostics to assess Parkinsonian disorders (
Table 1). Used in conjunction with other diagnostic tests, fluorodopa F-18 PET is primarily utilized to visualize dopaminergic nerve terminals in the striatum. Aromatic amino acid decarboxylase (AADC) of the striatum converts fluorodopa F-18 to fluorodopamine F-18. Monoamine oxidase (MAO) continues to break down fluorodopa F-18 to produce
18F. Urine removes about 80% of the radiation that is injected. Following intravenous dosing, this drug has a plasma half-life of between 1 and 3 h
[45].
2.4. Fluoroestradiol F-18
In May 2020, fluoroestradiol F-18 received FDA approval for use in patients with recurrent or metastatic breast cancer (
Table 1). Fluoroestradiol F-18 is a radioactive diagnostic agent used in PET imaging for the detection of estrogen receptor-positive lesions as a supplement to biopsy. The drug can be absorbed by any tumor cell that expresses estrogen receptors, including those that originate in the uterus or the ovaries. In vivo ER expression can be measured using
18F-fluoroestradiol PET, and its effectiveness as a predictive assay and indicator of in vivo pharmacodynamic response to endocrine therapy has been established in clinical studies. Only 10% of the total activity of fluoroestradiol F-18 at 2 h after delivery is related to the parent drug, which is highly extracted and degraded by the liver. Urinary and biliary excretion are the main methods of elimination. Unbound metabolites, primarily glucuronide and sulphate conjugates, are released in the bile, reabsorbed by enterohepatic circulation, and subsequently eliminated through the kidneys
[46].
2.5. Piflufolastat F-18
For the diagnosis of metastatic or recurring prostate cancer, piflufolastat F-18 is “a radiopharmaceutical diagnostic agent” used in PET to scan for PSMA-positive tumors (
Table 1). Although the most frequent non-cutaneous tumor affecting men in North America is prostate cancer, visualizing the size and location of tumor metastases and recurrences remains an obstacle in the treatment of this disease. While MRI and CT scans provide more detail, PET images are more sensitive and can detect malignant tissue in any part of the body. Piflufolastat
18F, commonly known as
18F-DCFPyL, is a radiopharmaceutical made of urea that binds to PSMA and makes malignant prostate tissue visible. Under the trade name Pylarify, it received FDA approval for the first time in May 2021 to enable earlier and more precise diagnosis of probable prostate cancer metastases
[47].
3. FDA-Approved Fluorinated Oligonucleotide Drugs
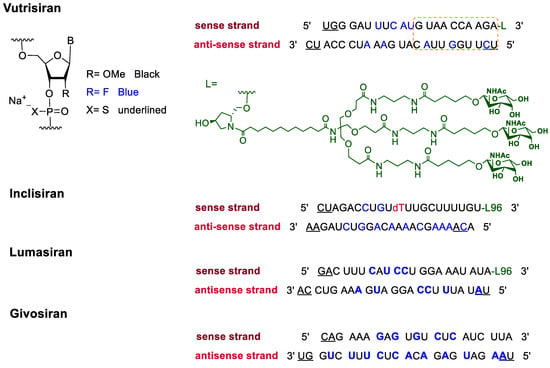
In addition to F-containing small molecules, in recent years, the FDA has approved (2019–2022) four oligonucleotides: vutrisiran (polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults); inclisiran (atherosclerotic cardiovascular disease and familial hypercholesterolemia); lumasiran (hyperoxaluria type 1); and givosiran (acute hepatic porphyria). These four drugs share a similar structure (
Figure 2): a double-stranded siRNA, with around 20 ribonucleosides for the sense and antisense strands (
Table 2). They have a total of six thiophosphate linkages, as well as several 2′-F-ribonucleoside units to improve the stability of the double strand
[48]. The remaining ribonucleosides are 2′-methoxy. With the same idea of increasing the stability, the 3′ end is linked to a short dendrimer of
N-acetylgalactosamine (GalNAc) to mediate the binding and internalization of the drug by hepatocytes
[49][50][51][49,50,51].
Figure 2.
Structures of fluorinated oligonucleotides.
Table 2.
FDA-Approved Fluorinated Oligonucleotide Drugs.
4. FDA-Approved Fluorinated Heterocyclic/Carbocyclic Drugs
The presence of numerous heterocyclic rings in natural products such as alkaloids, vitamins, antibiotics, peptides, etc., prompted the introduction of these motifs in synthetic pharmaceuticals
[52][53][52,53]. As a result, heterocycles are regarded important scaffolds for the synthesis of physiologically active molecules and potential drugs
[54][55][54,55]. Pyrrole, pyrrolidine, pyrrolidine-2,5-dione, imidazole, pyrazole, triazole, pyridine, piperidine, pyrimidine, pyrazine, triazine, quinoline, indole, and associated analogues are the most frequent heterocyclic cores found in bioactive compounds. Indeed, heterocyclic moieties are found in around 85% of bioactive chemicals
[56]. On the other hand, the inclusion of F atoms in pharmaceuticals introduced another essential tool for drug design in the second half of the twentieth century
[57][58][59][57,58,59].
Since the introduction of the first fluorocorticosteroid, namely fludrocortisone, in 1954
[9], the fluorinated drug market has grown exponentially, with these medicines accounting for 20% of those on the market, and approximately 30% of them becoming blockbusters, such as Lipitor, Fluoxetine, Linezolid, and Fluticasone
[59]. Over 300 fluorinated medicines have been approved for use as drugs to date
[57]. The success of the introduction of F atoms is linked to the peculiar physicochemical properties of the C–F bond
[60], namely its high bond strength, polarity, and minimal steric hindrance of F
[61], combined with general metabolic stability, which is still being studied
[62]. For example, the introduction of F allows researchers to readily modulate the pKa of neighboring functions, thereby enhancing bioavailability and affinity to certain receptors
[63][64][63,64].
Furthermore, due to the high electronegativity of F, monofluorination or trifluoromethylation of alkyl groups reduces the lipophilicity of drugs. Fluoro-arenes, on the other hand, are more lipophilic due to the reduced polarizability of the C–F bond
[65]. Moreover, the inclusion of an F atom improves membrane permeability
[66]. Fluorinated drugs are especially important because they are used as diagnostic tools in imaging procedures such as
19F-MRI and
18F-PET
[67][68][69][67,68,69]. Given the direct link between fluorinated moieties and heterocycles, a subclass of fluorinated heterocycles was established, which combines the strength of these two key scaffolds in modern medicinal chemistry. Fluorinated carbocyclic drugs are also available but these are in very small number as compared to fluorinated heterocyclic drugs.
The fluorinated heterocyclic/carbocyclic drugs approved by the FDA during 2016–2022 are reported in Table 3.
Table 3. FDA-Approved Fluorinated Heterocyclic/Carbocyclic Drugs.
FDA-Approved Fluorinated Heterocyclic/Carbocyclic Drugs.
| Name |
Structure |
Mode of Action |
References |
| Abemaciclib |
 |
CDK4/cyclin D1 complex inhibitor |
[70] |
| Alpelisib |
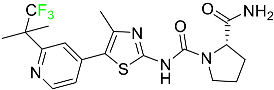 |
Phosphatidylinositol 3-kinase (PI3K) inhibitor |
[71] |
| Atogepant |
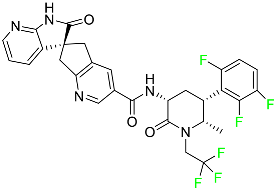 |
CGRP receptor antagonist |
[72] |
| Apalutamide |
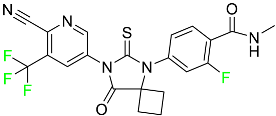 |
Androgen receptor inhibitor |
[73] |
| Asciminib |
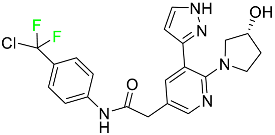 |
ABL/BCR–ABL1 tyrosine kinase inhibitor |
[74] |
| Avacopan |
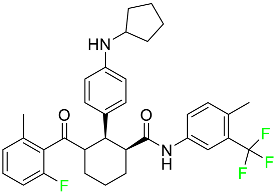 |
5a receptor (C5aR) antagonist |
[75] |
| Avapritinib |
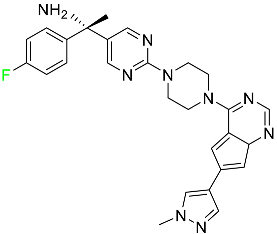 |
Selective tyrosine kinase inhibitor |
[76] |
| Baloxavir marboxil |
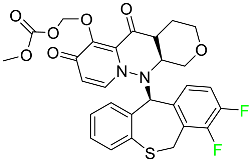 |
Endonuclease inhibitor |
[77] |
| Belzutifan |
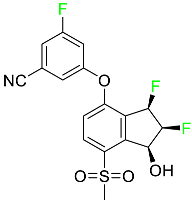 |
Hypoxia-inducible factor 2 inhibitor |
[78] |
| Berotralstat |
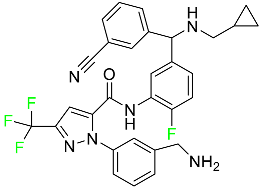 |
Plasma kallikrein inhibitor |
[79] |
| Bictegravir |
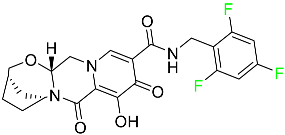 |
Integrase inhibitor |
[80] |
| Binimetinib |
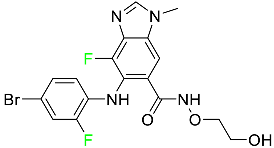 |
MEK inhibitor |
[81] |
| Cabotegravir |
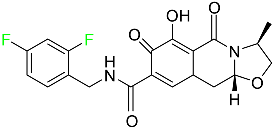 |
HIV-1 integrase inhibitor |
[82] |
| Capmatinib |
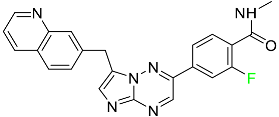 |
Kinase inhibitor |
[83] |
| Dacomitinib |
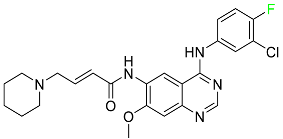 |
EGFR protein inhibitor, including EGFR/HER1, HER2, and HER4 |
[84] |
| Delafloxacin |
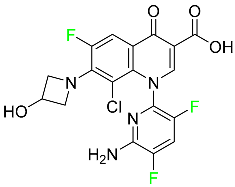 |
Bacterial DNA gyrase and topoisomerase IV inhibitor |
[85] |
| Doravirine |
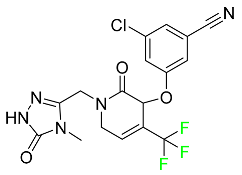 |
Non-nucleoside reverse transcriptase inhibitor |
[86] |
| Elagolix |
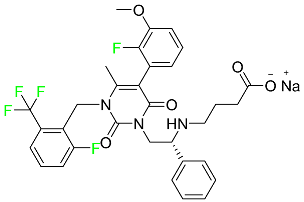 |
Gonadotropin-releasing hormone receptor antagonist (GnRH) |
[87] |
| Elexacaftor |
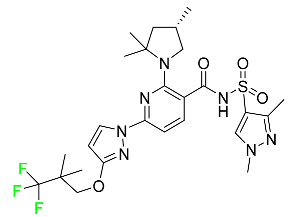 |
CFTR corrector |
[88] |
| Emtricitabine |
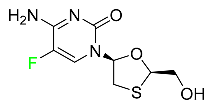 |
Nucleoside reverse transcriptase inhibitor (NRTI) |
[89] |
| Enasidenib |
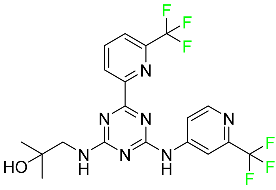 |
Isocitrate dehydrogenase-2 inhibitor |
[90] |
| Encorafenib |
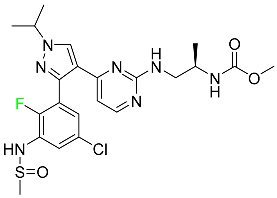 |
Kinase inhibitor |
[91] |
| Eravacycline |
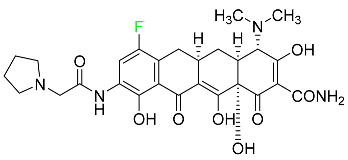 |
Binds to the bacterial ribosomal 30S subunit |
[92] |
| Fostamatinib |
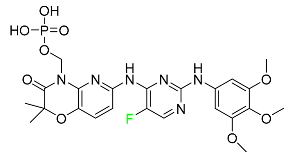 |
Tyrosine kinase inhibitor |
[93] |
| Glecaprevir |
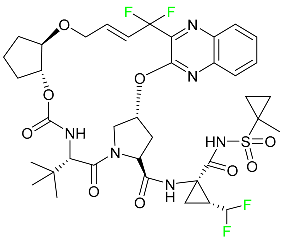 |
NS3/4A and NS5A inhibitor |
[94] |
| Ivosidenib |
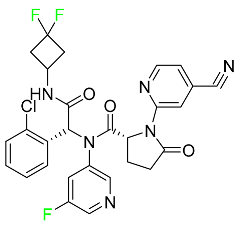 |
Isocitrate dehydrogenase-1 (IDH1) inhibitor |
[95] |
| Larotrectinib |
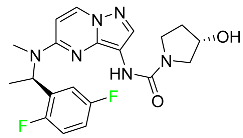 |
Tropomyosin receptor kinase (Trk) inhibitor |
[96] |
| Lasmiditan |
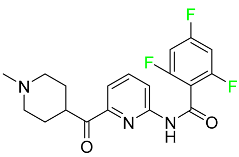 |
High-affinity serotonin (5-HT) 1F receptor agonist |
[87] |
| Lemborexant |
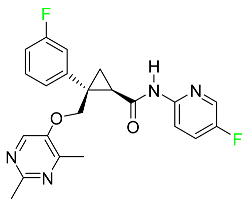 |
OX1R and OX2R antagonist |
[97] |
| Letermovir |
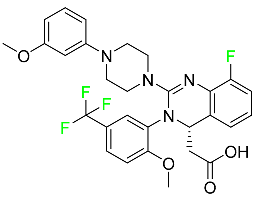 |
DNA terminase CMV complex inhibitor |
[98] |
| Lorlatinib |
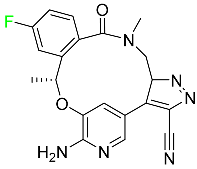 |
Tyrosine kinase inhibitor |
[99] |
| Lumateperone |
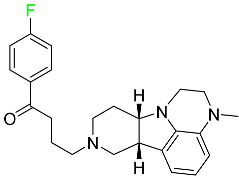 |
Receptor antagonist of 5-HT2A receptor and antagonist of several dopamine receptors (D1, D2, and D4) |
[100] |
| Melphalan flufenamide |
 |
Peptidase-enhanced cytotoxic (PEnC) |
[101] |
| Netupitant |
 |
Selective neurokinin 1 (NK1) receptor antagonist |
[100] |
| Osilodrostat |
 |
Cortisol synthesis inhibitor |
[102] |
| Pexidartinib |
 |
Tyrosine kinase inhibitor |
[103] |
| Pibrentasvir |
 |
NS3/4A and an NS5A inhibitor |
[94] |
| Pralsetinib |
 |
Kinase inhibitor |
[104] |
| Pretomanid |
 |
Inhibitor of cell wall biosynthesis via blockage of the oxidation of hydroxymycolate to ketomycolate |
[72] |
| Pimavanserin |
 |
Serotonin 5-HT2A receptor antagonist |
[105] |
| Relugolix |
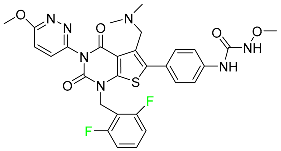 |
Nonpeptide GnRH receptor antagonist |
[106] |
| Ripretinib |
 |
Protein kinase inhibitor |
[107] |
| Rucaparib |
 |
Polymerase inhibitor |
[108] |
| Selinexor |
 |
Selective nuclear transport (SINE) inhibitor |
[109] |
| Selumetinib |
 |
Selective MEK 1 and MEK 2 inhibitor |
[110] |
| Siponimod |
 |
Sphingosine 1-phosphate receptor (S1P) modulator |
[111] |
| Sofosbuvir |
 |
Hepatitis C NS5B protein inhibitor |
[112] |
| Sotorasib |
 |
KRAS G12C inhibitor |
[113] |
| Tafenoquine |
 |
Antiparasitic agent |
[114] |
| Talazoparib |
 |
PARP1/2 enzyme inhibitor |
[115] |
| Tecovirimat |
 |
Orthopoxvirus VP37 inhibitor |
[116] |
| Telotristat Ethyl |
 |
Tryptophan hydroxylase inhibitor |
[117] |
| Tezacaftor |
 |
CFTR potentiator |
[88] |
| Ubrogepant |
 |
CGRP antagonist |
[114] |
| Umbralisib |
 |
Kinase inhibitor |
[118] |
| Upadacitinib |
 |
Selective Janus kinase-1 (JAK-1) inhibitor |
[119] |
| Vericiguat |
 |
Guanylate cyclase (sGC) direct stimulant |
[120] |
| Voxilaprevir |
 |
Protease inhibitor |
[121] |