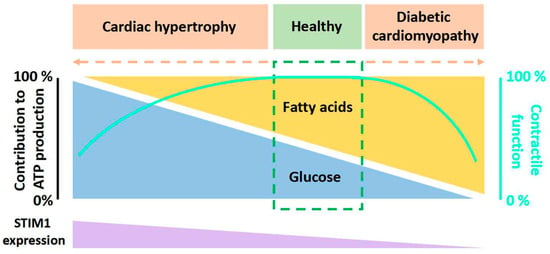
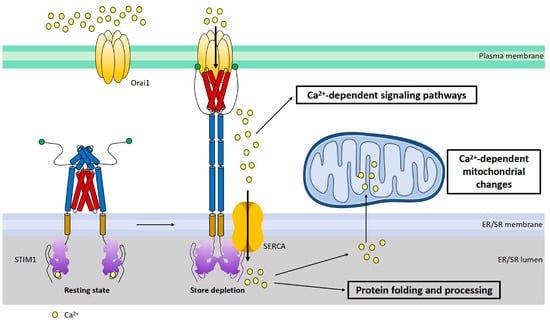
Figure 3. Cartoon illustration of activation and function of STIM1. STIM1 is a single transmembrane protein resident in ER/SR. Orai1 channel functions as a hexamer on plasma membrane. Under the resting state, the cytosolic region of STIM1 maintains an inactive folding configuration. Upon SR Ca
2+ store depletion, the loss of Ca
2+ triggers the dimerization of the STIM1 EF-SAM domain in the ER lumen, leading to the uncaging of its SOAR domain in the cytosol. SOAR would then bind and open Orai1, the pore-forming protein, inducing store-operated Ca
2+ entry (SOCE). In addition to its critical roles in mediating many Ca
2+-dependent signaling pathways, SOCE is also crucial for the maintenance of Ca
2+ homeostasis within the ER/SR and mitochondria. Proper-sized ER Ca
2+ store is essential for correct protein folding and processing as well as for maintaining Ca
2+-dependent mitochondrial changes of the cell.
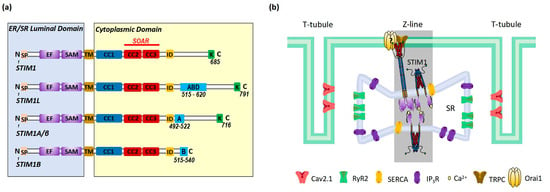
There are at least three known alternative splicing variants of STIM1 (STIM1L, STIM1A/β, and STIM1B) (
Figure 2a). STIM1A/β has an insertion of 31-amino-acid peptide after the SOAR in the cytoplasmic domain, which is found in astrocytes, heart, kidney, and testes
[17][45]. While STIM1A/β has been shown to more efficiently mediate SOCE with faster kinetics by disordering the cytosolic inhibitory domain in HEK, HeLa and glioblastoma cells
[18][46], it was also shown to reduce SOCE in astrocytes in heterologous expression while at the same time increasing NFAT translocation due to a more efficient recruitment of the NFAT signalosome
[17][45]. Indeed, it is possible that differential NFAT activation and not small differences in SOCE mediate a splice variant-specific effect in cells or else that the functions of STIM1A/β may differ in different cell types. STIM1B is a short isoform of STIM1, which lacks 170 amino acids but has an extra peptide of 26 amino acids in the cytoplasmic domain. STIM1B is a neuron-specific variant and induces slower I
CRAC and the inactivation of I
CRAC [19][47]. The C-terminal end of STIM1L bears an extra 106-amino-acids-long peptide that contains an actin-binding domain (ABD); thus, it can facilitate rapid SOCE
[20][21][48,49]. STIM1L is only expressed in skeletal muscle cells, neonatal cardiomyocytes or hypertrophic adult cardiomyocytes
[22][24].
In non-excitable cells, STIM1 is distributed throughout the ER at rest. Upon store depletion, STIM1 translocates to ER–PM junctions to interact and activate Orai1 to induce SOCE
[9][10] (
Figure 3). In cardiomyocytes, STIM1 is mostly localized in the SR at the Z-lines and is believed to function as a sensor for SR store
[23][24][25][26][16,17,19,21]; the underlying mechanisms for this uneven distribution still awaits further investigation. Orai1 is detected in the sarcolemma membranes (
Figure 2b). Around the area where STIM1 and Orai1 are resident is the well-known diad, which is a key place for excitation–contraction coupling (ECC) mediated by ryanodine receptor 2 (RyR2) at the terminal cisternae of the SR and voltage-gated Ca
2+ channel Ca
v2.1 at the T-tubule (
Figure 2b). Additionally, SR Ca
2+ ATPase (SERCA) and inositol-1,4,5-triphosphate receptor (IP
3R) are also involved in Ca
2+ handling in this area (
Figure 2b)
[27][50]. Different from non-excitable cells, STIM1 forms constitutive puncta near sarcolemma and does not change its distribution upon store depletion
[28][25]. In addition, there is barely co-localization between STIM1 and Orai1 in cardiomyocytes
[28][25], indicating fewer involvements of Orai1 in STIM1-mediated SOCE. Indeed, STIM1 can hardly induce classic SOCE with Orai1, which is characterized by highly calcium-selective I
CRAC with large inward rectification and very positive reversal potential
[28][29][25,51]. Instead, a non-selective current is more commonly induced by STIM1 likely via the activation of transient receptor potential channels such as TRPC
[29][30][31][51,52,53]. However, it is still controversial whether TRPC channels can be directly activated by STIM1
[32][54]. And Orai2 and Orai3 are both expressed in cardiac cells
[33][23], thus it is also likely that STIM1 might mediate SOCE via interactions with Orai2 or Orai3.
STIM1-mediated SOCE is crucial for refilling ER Ca
2+ [9][34][35][10,55,56] (
Figure 3), as STIM1 deletion blocked the fast refilling of ER Ca
2+ after store depletion
[36][37][57,58]. Also, a decrease in STIM1 expression impaired ER Ca
2+ refilling, which could be restored by STIM1 overexpression
[38][30]. Similarly, it is believed that one key function of STIM1-mediated SOCE in cardiomyocytes is to refill SR Ca
2+ and thereby maintain SR Ca
2+ homeostasis for proper protein folding and processing
[39][59]. STIM1 knockdown by siRNA in cultured neonatal rat ventricular cardiomyocytes reduced the SR Ca
2+ content
[23][16]. In addition, it has been shown that STIM1 overexpression increased the SR Ca
2+ level in rat ventricular myocytes
[30][52]. However, a study from Correll and colleagues showed that STIM1 overexpression had no effect on the total SR Ca
2+ load in mouse ventricular myocytes. They suggested that elevated STIM1 resulted in increased Ca
2+ uptake into the SR but also RyR2-dependent Ca
2+ leak; therefore, the total SR Ca
2+ load remained unaltered
[25][19]. Ca
2+ influx through STIM1-mediated SOCE refills ER/SR Ca
2+ by SERCA. SERCA pumps have been shown to co-localize with STIM1 in different cell types, coupling Ca
2+ entry with Ca
2+ refilling
[35][56]. In cardiomyocytes, SERCA is mainly resident at Z-lines where STIM1 is localized
[40][60] (
Figure 2b), likely enabling the similar function of Ca
2+ entry–Ca
2+ refilling coupling. A disruption of ER Ca
2+ homeostasis can cause ER stress, leading to the accumulation of unfolded proteins and thus unfolded protein response (UPR)
[41][61]. STIM1-knockout hearts displayed increased levels of ER stress marker CHOP
[39][59]. Single-nucleotide polymorphisms (SNPs) in the STIM1 gene are correlated with ER stress in patients undergoing cardiac catheterization and spontaneous hypertension in rats
[42][43][62,63]. Since ER stress and UPR often correlate with metabolic disorders
[44][45][46][47][48][64,65,66,67,68], it is likely that STIM1 may mediate an energy substrate preference in cardiac metabolic diseases through regulating ER Ca
2+ homeostasis.
In addition to refilling SR Ca
2+, STIM1 could also activate downstream Ca
2+-dependent metabolic pathways (
Figure 3). It is widely accepted that Ca
2+ signaling regulates cardiac energy metabolism through the direct activation of Ca
2+-dependent proteins, kinases, enzymes and transcriptional regulators
[49][50][69,70]. For instance, pyruvate dehydrogenase (PDH), PDH kinase (PDK), AMP-activated protein kinase (AMPK) and AKT are involved in the regulation of cardiac energy metabolism and are mediated by Ca
2+ [51][52][53][71,72,73]. In addition, Ca
2+ signaling regulates glycolysis and glucose oxidation in the heart and is crucial for the translocation of glucose transporter (GLUT) 4 and FA transporter CD36 (also known as the scavenger receptor B2, SR-B2) to the sarcolemma to induce cardiac glucose and FA uptake, respectively
[54][55][74,75]. Moreover, peroxisome proliferator-activated receptor (PPAR) and PPARγ coactivator-1α (PGC-1α), key transcriptional regulators of expression of genes encoding mitochondrial oxidation enzymes, are associated with Ca
2+ signaling as well
[56][57][76,77]. Considering the key role of STIM1 in SOCE, STIM1 may regulate the energy substrate preference in cardiac metabolic diseases through Ca
2+ signaling. Moreover, STIM1 can directly interact with a variety of other proteins
[30][36][58][59][60][61][62][63][64][52,57,78,79,80,81,82,83,84], and alternative splicing variants of STIM1 might have splice-specific partners
[17][45]. Interestingly, a recent report has shown that STIM1 negatively regulates the activation of stimulator of interferon genes (STING) through tethering STING to ER
[65][85]. Of note, the cyclic GMP–AMP synthase (gGAS)–STING signaling pathway mediates cardiac metabolic abnormalities
[66][86]. Therefore, STIM1 may regulate the energy substrate preference in cardiac metabolic diseases through the gGAS–STING signaling pathway or similarly via direct interactions with some other proteins correlating to energy metabolism. Given that glycolytic enzymes are clustered near SR
[67][87] where STIM1 is resident, the likelihood of the conjecture increases.
Moreover, STIM1-mediated SOCE could contribute to cardiac metabolic diseases via the alteration of mitochondrial Ca
2+ homeostasis or fission. Mitochondrial function including ATP production and reactive oxygen species (ROS) generation is regulated by Ca
2+ [68][69][88,89]. Mitochondria takes up Ca
2+ via voltage-dependent anion channel (VDAC) and mitochondrial Ca
2+ uniporter (MCU)
[70][71][72][73][90,91,92,93]. Normally, the ER/SR transmits Ca
2+ to mitochondria to regulate mitochondrial function
[74][94]. Therefore, STIM1-mediated SOCE is related to mitochondrial function through involvement in mitochondrial Ca
2+ uptake from the ER/SR
[25][75][76][77][78][19,95,96,97,98] (
Figure 3). Alterations of ER/SR Ca
2+ homeostasis due to the changes of STIM1-mediated SOCE would affect ER/SR–mitochondrial Ca
2+ communication and thereby cause mitochondrial dysfunction. Excessive or insufficient mitochondrial Ca
2+ in cardiomyocytes leads to mitochondrial dysfunction
[79][99], which is associated with cardiac metabolic diseases such as cardiac hypertrophy and diabetic cardiomyopathy
[79][80][81][82][99,100,101,102]. Interestingly, research has shown a decrease in mitochondrial Ca
2+ uptake in diabetic hearts and restoring mitochondrial Ca
2+ rescued mitochondrial and cardiac dysfunction
[83][103]. In addition, Ca
2+–calcineurin is involved in the regulation of mitochondrial fission, which is associated with the development of both cardiac hypertrophy and diabetic cardiomyopathy
[78][84][85][86][87][88][89][90][98,104,105,106,107,108,109,110]. A recent study showed that STIM1 deficiency in cardiomyocytes changed mitochondrial morphology, which is indicative of an elevation of mitochondrial fission
[39][91][32,59]. This phenomenon is in line with the fact that pro-fusion proteins (optic atrophy 1, Opa1; mitofusion 2, Mfn2) are downregulated, and the pro-fission protein (dynamin-related protein 1, Drp1) is activated, promoting mitochondrial fission in diabetic cardiomyocytes
[78][88][89][90][98,108,109,110]. Notably, Mfn2 positively regulates SOCE via mediating STIM1 movement to ER–PM junctions
[92][111], indicating there is a feedback loop between mitochondrial dynamics and STIM1-mediated SOCE. Therefore, any increase or decrease in STIM1 could induce mitochondrial dysfunction via influencing the mitochondrial Ca
2+ level and mitochondrial fission, resulting in the alterations in cardiac glucose and FA oxidation. In turn, increased ROS due to mitochondrial dysfunction could downregulate STIM1 expression through NF-kB as a feedback effect
[93][112]. Moreover, a study of T cells has shown that STIM1 deletion resulted in the downregulated expression of many subunits of the electron transport chain (ETC), suggesting that STIM1 is involved in mitochondrial function by controlling the expression of subunits of the ETC
[94][113]. It would be intriguing to test this also the case in cardiomyocytes.
Thank you for your interest. More content can be found in the original paper.