Chronic disorders are strongly linked to cardiovascular (CV) diseases, and it is unanimously accepted that regular exercise training is a key tool to improving CV risk factors, including diabetes, dyslipidemia, and obesity. Increased oxidative stress due to an imbalance between reactive oxygen species production and their scavenging by endogenous antioxidant capacity is the common ground among these metabolic disorders, and each of them affects platelet function. Habitual physical exercise triggers important mechanisms related to the exercise benefits for health improvement and protects against CV events. Platelets play an important role in many physiological and pathophysiological processes, including the development of arterial thrombosis, and physical (in)activity has been shown to interfere with platelet function. Although data reported by studies carried out on this topic show discrepancies, knowledge on platelet function affected by exercise mainly depends on the type of applied exercise intensity and whether acute or habitual, strenuous or moderate, thus suggesting that physical activity and exercise intensity may interfere with platelet function differently.
- platelets
- exercise
- obesity
- dyslipidemia
- diabetes
1. Introduction
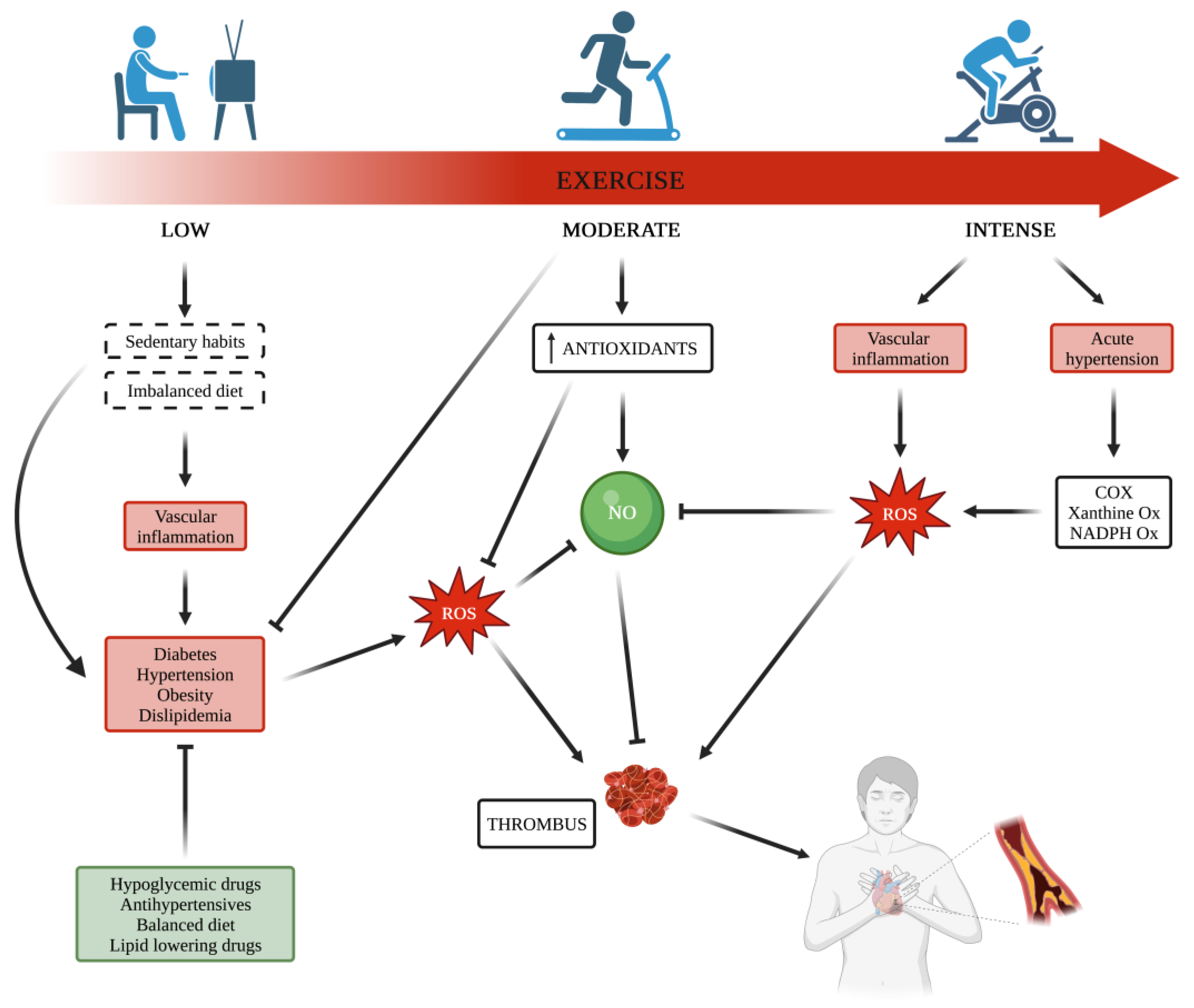
2. Metabolic Diseases and Exercise Effects on Platelets
| Study Design | Population | Number of Individuals | Exercise Protocol | Platelet Parameters | Effects of Exercise | Reference |
|---|---|---|---|---|---|---|
| Obesity | ||||||
| Randomized controlled trial | Men | 53 | Progressive training program for 12 weeks | ADP-induced platelet aggregation | ↓ ADP-induced PLT aggregation (Ex group vs. Ref group) | [135][63] |
| Exercise group: 26 | Sessions: five times/week, 45 to 60 min per session. | Release of ATP | No significant change | |||
| Reference group: 27 | Serum TXB2 | ↓ TXB2 (Ex group vs. Ref group) | ||||
| Prospective randomized trial | CAD | 46 Cases CR: 21 standard cardiac rehab (CR) Cases HCR: 25 high caloric CR |
All: 4 months of intervention + 1 month of weight stabilization with training CR: 3 sessions/week of 25 to 40 min HCR: 5 to 7 sessions/week of 45 to 60 min |
P-Selectin expression GPIIb/IIIa activation |
↓ P-Selectin expression (5 months vs. baseline, all subjects) ↓ P-Selectin expression (5 months vs. baseline, HCR) GPIIb-IIIa activation no significant change (5 months vs. baseline, all subjects) |
[189][64] |
| Controlled clinical intervention study | Women | 42 | All: 30-min walking exercise test with an intensity of 70% of individual peak VO2 | Thrombus formation Collagen-induced platelet aggregation Platelet adhesion |
↑ clot formation time (post vs. pre) ↓ alpha-angle (post vs. pre) No significant change |
[121][65] |
| PLT count | No significant change | |||||
| Case-control study | 23 | All: in two different days two exercise protocols | PLT EVs (CD61+) | ↓ PLT EVs (postexercise 24 h vs. basal, all subjects) | [190][67] | |
| Cases: 15 | Pilot test: incremental exercise on a treadmill until voluntary exhaustion | |||||
| Controls: 8 | Submaximal test: 30 min of moderate constant workload | |||||
| Randomized controlled trial | Rats | 24 | All: 15 weeks | ADP-induced platelet aggregation | ↓ ADP-induced PLT aggregation (HCFD + Ex vs. HCFD) | [191][68] |
| Cases Ex: 6 | Group Ex: swimming 3 days/week, 1 h | PLT count | No significant change | |||
| Cases HCFD: 6 | Group HCFD: high-fat diet | |||||
| Cases HCFD + Ex: 6 | Group HCFD + Ex: high-fat diet + swimming from the 11th week to the 15th week | |||||
| Controls: 6 | Controls: no exercise training | |||||
| Lipid Profile Alterations |
||||||
| Randomized controlled trial | Mice | 63 | All: 8 weeks | PLT aggregation rate | ↓ PLT aggregation rate (FE vs. HF) | [186][60] |
| Controls: 21 | Cases HF: high fat diet | PLT spread on fibrinogen | ↓ PLT spread on fibrinogen (FE vs. HF) | |||
| Cases HF: 21 | Cases FE: swimming 60 min/day, 5 days/week | PLT pAKT level | ↓ PLT pAKT level (FE vs. HF) | |||
| Cases FE: 21 | ||||||
| Randomized controlled trial | Sedentariety | 10 | 1. Strenuous, acute exercise | ADP-induced PLT aggregation | ↑ ADP-induced PLT aggregation (strenous ex vs. rest) | [75][106] |
| 2. Ergometer cycling: | ADP-induced [Ca2+]i elevation | ↑ ADP-induced [Ca2+]i elevation (strenous ex vs. rest) | ||||
| 30 min/day, 5 days/week, 8 weeks | ox-LDL-induced PLT aggregation | ↑ ox-LDL-induced PLT aggregation (strenous ex vs. rest) | ||||
| 3. 12 Weeks detraining | ox-LDL-induced [Ca2+]i elevation | ↑ ox-LDL-induced [Ca2+]i elevation (strenous ex vs. rest) | ||||
| ↓ ADP-induced PLT aggregation (training vs. pre-training) | ||||||
| ↓ ADP-induced [Ca2+]i elevation (training vs. pre-training) | ||||||
| ↓ ox-LDL-induced PLT aggregation (training vs. pre-training) | ||||||
| ↓ ox-LDL-induced [Ca2+]i elevation (training vs. pre-training) | ||||||
| ↑ ADP-induced PLT aggregation (detraining vs. training) | ||||||
| ↑ ADP-induced [Ca2+]i elevation (detraining vs. training) | ||||||
| ↑ ox-LDL-induced PLT aggregation (detraining vs. training) | ||||||
| ↑ ox-LDL-induced [Ca2+]i elevation (detraining vs. training) | ||||||
| Randomized controlled trial | Healthy | 30 | Treadmill test using Bruce protocol | Plasma TX levels | ↑ plasma TX levels (pre vs. post) | [84][105] |
| Plasma PGI2 levels | No significant change | |||||
| After ex vivo addition of mildly ox-LDL: | ||||||
| TX release | ↓ TX release (mildly ox-LDL+ vs. mildly ox-LDL-) | |||||
| Collagen-induced PLT aggregability | ↓ collagen-induced PLT aggregability | |||||
| (mildly ox-LDL+ vs. mildly ox-LDL-) | ||||||
| Cross-sectional study | CHD | 18 | All: 8 weeks | Urinary 8-iso-PGF2a | ↓ Urinary 8-iso-PGF2a (post vs. pre) | [219][99] |
| Sedentariety | 2 sessions/week on cycle ergometer | Urinary 11-dehydro-TXB2 | ↓ Urinary 11-dehydro-TXB2 (post vs. pre) | |||
| Low HDL-c | 55 min/session, supervised | |||||
| DIABETES | ||||||
| Randomized controlled trial | 24 | All: 12 weeks | ADP-induced PLT aggregation | ↓ ADP-induced PLT aggregation (Cases vs. Controls) | [240][122] | |
| Cases: 12 | Cases: walking and running on the treadmill in Non-consecutive days |
miRNA-223 expression | ↑ miRNA 223 expression (Cases vs. Controls) | |||
| Controls: 12 | Controls: no exercise training | |||||
| Quasi-experimental controlled trial | Sedentariety | 24 | All: 8 weeks | Collagen-induced PLT aggregation | ↓ collagen-induced PLT aggregation (post vs. pre, cases) | [239][121] |
| Cases: mean intensity treadmill, 3 times/week | PLT, MPV, PDW, PCT | ↓ MPV, ↓ PDW (post vs. pre, cases) | ||||
| Controls: no exercise training | Glycoprotein IIb (GPIIb) receptor expression | Down-regulated (post vs. pre, cases) | ||||
| miR-130a expression | No significant change | |||||
| Randomized controlled trial | CAD | 74 | All: 12 months | PMVs | No significant change | [242][124] |
| Albuminuria (n = 25) | Cases: 38 | Cases: aerobic and resistance training | PMVs in patients with albuminuria | ↓ PMVs carrying TF (CD61+/CD142+) (post vs. pre, cases) | ||
| Controls: 36 | 150 min/week | ↓ PMVs carrying vWF (CD31+/CD42b) (post vs. pre, cases) | ||||
| Prospective study | Aspirin treatment | 79 | All: single treadmill exercise test | PLT aggregation (ASPI test) | Cases: ↑↑ PLT aggregation (post vs. pre) | [243][125] |
| Cases: T2DM | Cases: 43 | Controls: ↑ PLT aggregation (post vs. pre) | ||||
| Controls: no T2DM | Controls: 36 | |||||
| Randomized crossover study | 11 | All: post-meal walks | PMVs | No significant change | [245][127] | |
| 15 min/day | MPAs | ↑ MPAs (post vs. pre, cases) | ||||
| 4 days | ||||||
| Randomized controlled trial | Cases: T2DM | 16 | 1 session of nuclear exercise stress test | PLT count | No significant change | [249][131] |
| Controls: no T2DM | Cases: 8 | Bruce protocol | ADP-induced PLT activation | No significant change | ||
| Controls: 8 | Collagen-induced PLT activation | No significant change | ||||
| Arachidonic acid-induced PLT activation | No significant change | |||||
| Aspirin responsiveness | No significant change | |||||
| Randomized controlled trial | Women | 20 | All: 8 weeks | ADP-induced platelet aggregation | ↓ ADP-induced PLT aggregation (Cases vs. Controls) trend p = 0.06 | [241][123] |
| Cases: 10 | Cases: Endurance training, 3 non-consecutive days/week | miRNA-223 expression | ↑ miRNA 223 expression (Cases vs. Controls) trend p = 0.06 | |||
| Controls: 10 | Controls: no exercise training | |||||
| Randomized crossover study | Women | 15 | All: 2 resistance exercises | P-Selectin expression | No significant change | [250][132] |
| Cases: training with blood-flow restriction (BFR) | GPIIb/IIIa activation CD42 expression |
No significant change No significant change |
||||
| Controls: training w/o BFR | CD61 expression | ↓ CD61 expression (Controls post vs. pre) | ||||
| PLT count | ↑ PLT count (Cases vs. Controls) | |||||
| Plateletcrit (PCT) | ↑ PCT (Controls vs. Cases) | |||||
| PDW | No significant change | |||||
| MPV | No significant change |
References
- Heinonen, I.; Kalliokoski, K.K.; Hannukainen, J.C.; Duncker, D.J.; Nuutila, P.; Knuuti, J. Organ-specific physiological responses to acute physical exercise and long-term training in humans. Physiology 2014, 29, 421–436.
- Pedersen, B.K.; Saltin, B. Exercise as medicine-evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. S3), 1–72.
- Sharma, S.; Pelliccia, A.; Gati, S. The “Ten Commandments” for the 2020 ESC Guidelines on Sports Cardiology and Exercise in Patients with Cardiovascular Disease. Eur. Heart J. 2021, 42, 6–7.
- Dibben, G.; Faulkner, J.; Oldridge, N.; Rees, K.; Thompson, D.R.; Zwisler, A.-D.; Taylor, R.S. Exercise-based cardiac rehabilitation for coronary heart disease. Cochrane Database Syst. Rev. 2021, 11, CD001800.
- Alves, A.J.; Wu, Y.; Lopes, S.; Ribeiro, F.; Pescatello, L.S. Exercise to Treat Hypertension: Late Breaking News on Exercise Prescriptions That FITT. Curr. Sports Med. Rep. 2022, 21, 280–288.
- Doewes, R.I.; Gharibian, G.; Zadeh, F.A.; Zaman, B.A.; Vahdat, S.; Akhavan-Sigari, R. An Updated Systematic Review on the Effects of Aerobic Exercise on Human Blood Lipid Profile. Curr. Probl. Cardiol. 2023, 48, 101108.
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623.
- Scheen, A.J. Cardiovascular Effects of New Oral Glucose-Lowering Agents: DPP-4 and SGLT-2 Inhibitors. Circ. Res. 2018, 122, 1439–1459.
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291.
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. Biomed. Res. Int. 2018, 2018, 6508709.
- Russo, I.; Traversa, M.; Bonomo, K.; De Salve, A.; Mattiello, L.; Del Mese, P.; Doronzo, G.; Cavalot, F.; Trovati, M.; Anfossi, G. In central obesity, weight loss restores platelet sensitivity to nitric oxide and prostacyclin. Obesity 2010, 18, 788–797.
- Barale, C.; Cavalot, F.; Frascaroli, C.; Bonomo, K.; Morotti, A.; Guerrasio, A.; Russo, I. Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. Int. J. Mol. Sci. 2020, 21, 4983.
- Förstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010, 459, 923–939.
- Ikarugi, H.; Shibata, M.; Shibata, S.; Ishii, H.; Taka, T.; Yamamoto, J. High intensity exercise enhances platelet reactivity to shear stress and coagulation during and after exercise. Pathophysiol. Haemost. Thromb. 2003, 33, 127–133.
- Sharma, S.; Merghani, A.; Mont, L. Exercise and the heart: The good, the bad, and the ugly. Eur. Heart J. 2015, 36, 1445–1453.
- Després, J.P. Intra-abdominal obesity: An untreated risk factor for Type 2 diabetes and cardiovascular disease. J. Endocrinol. Investig. 2006, 29, 77–82.
- Davì, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494.
- Davì, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014.
- Bird, S.R.; Hawley, J.A. Update on the effects of physical activity on insulin sensitivity in humans. BMJ Open Sport. Exerc. Med. 2016, 2, e000143.
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 2018, 5, 135.
- Black, S.E.; Mitchell, E.; Freedson, P.S.; Chipkin, S.R.; Braun, B. Improved insulin action following short-term exercise training: Role of energy and carbohydrate balance. J. Appl. Physiol. 2005, 99, 2285–2293.
- Newsom, S.A.; Everett, A.C.; Hinko, A.; Horowitz, J.F. A single session of low-intensity exercise is sufficient to enhance insulin sensitivity into the next day in obese adults. Diabetes Care 2013, 36, 2516–2522.
- Heiston, E.M.; Liu, Z.; Ballantyne, A.; Kranz, S.; Malin, S.K. A single bout of exercise improves vascular insulin sensitivity in adults with obesity. Obesity 2021, 29, 1487–1496.
- Falcon, C.; Pfliegler, G.; Deckmyn, H.; Vermylen, J. The platelet insulin receptor: Detection, partial characterization, and search for a function. Biochem. Biophys. Res. Commun. 1988, 157, 1190–1196.
- López-Aparicio, P.; Rascón, A.; Manganiello, V.C.; Andersson, K.E.; Belfrage, P.; Degerman, E. Insulin induced phosphorylation and activation of the cGMP-inhibited cAMP phosphodiesterase in human platelets. Biochem. Biophys. Res. Commun. 1992, 186, 517–523.
- Anfossi, G.; Russo, I.; Trovati, M. Platelet resistance to the anti-aggregating agents in the insulin resistant states. Curr. Diabetes Rev. 2006, 2, 409–430.
- Ferreira, I.A.; Mocking, A.I.M.; Feijge, M.A.H.; Gorter, G.; van Haeften, T.W.; Heemskerk, J.W.M.; Akkerman, J.-W.N. Platelet inhibition by insulin is absent in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 417–422.
- Mendis, S. The contribution of the Framingham Heart Study to the prevention of cardiovascular disease: A global perspective. Prog. Cardiovasc. Dis. 2010, 53, 10–14.
- Lakka, H.-M.; Laaksonen, D.E.; Lakka, T.A.; Niskanen, L.K.; Kumpusalo, E.; Tuomilehto, J.; Salonen, J.T. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA 2002, 288, 2709–2716.
- Mente, A.; Yusuf, S.; Islam, S.; McQueen, M.J.; Tanomsup, S.; Onen, C.L.; Rangarajan, S.; Gerstein, H.C.; Anand, S.S. INTERHEART Investigators Metabolic syndrome and risk of acute myocardial infarction a case-control study of 26,903 subjects from 52 countries. J. Am. Coll. Cardiol. 2010, 55, 2390–2398.
- Novo, S.; Peritore, A.; Guarneri, F.P.; Corrado, E.; Macaione, F.; Evola, S.; Novo, G. Metabolic syndrome (MetS) predicts cardio and cerebrovascular events in a twenty years follow-up. A prospective study. Atherosclerosis 2012, 223, 468–472.
- Towfighi, A.; Ovbiagele, B. Metabolic syndrome and stroke. Curr. Diab Rep. 2008, 8, 37–41.
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880.
- Després, J.-P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887.
- Kakafika, A.I.; Liberopoulos, E.N.; Karagiannis, A.; Athyros, V.G.; Mikhailidis, D.P. Dyslipidaemia, hypercoagulability and the metabolic syndrome. Curr. Vasc. Pharmacol. 2006, 4, 175–183.
- Ritchie, S.A.; Connell, J.M.C. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 319–326.
- Mertens, I.; Van Gaal, L.F. Obesity, haemostasis and the fibrinolytic system. Obes. Rev. 2002, 3, 85–101.
- Anfossi, G.; Russo, I.; Trovati, M. Platelet dysfunction in central obesity. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 440–449.
- Pignatelli, P.; Menichelli, D.; Pastori, D.; Violi, F. Oxidative stress and cardiovascular disease: New insights. Kardiol. Pol. 2018, 76, 713–722.
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341.
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The metabolic syndrome and antioxidant concentrations: Findings from the Third National Health and Nutrition Examination Survey. Diabetes 2003, 52, 2346–2352.
- Patti, M.-E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395.
- Fetterman, J.L.; Holbrook, M.; Westbrook, D.G.; Brown, J.A.; Feeley, K.P.; Bretón-Romero, R.; Linder, E.A.; Berk, B.D.; Weisbrod, R.M.; Widlansky, M.E.; et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 53.
- Yetkin, E. Mean platelet volume not so far from being a routine diagnostic and prognostic measurement. Thromb. Haemost. 2008, 100, 3–4.
- Coban, E.; Ozdogan, M.; Yazicioglu, G.; Akcit, F. The mean platelet volume in patients with obesity. Int. J. Clin. Pract. 2005, 59, 981–982.
- Pinto, R.V.L.; Rodrigues, G.; Simões, R.L.; Porto, L.C. Analysis of Post-Sample Collection EDTA Effects on Mean Platelet Volume Values in Relation to Overweight and Obese Patient Status. Acta Haematol. 2019, 142, 149–153.
- Montilla, M.; Santi, M.J.; Carrozas, M.A.; Ruiz, F.A. Biomarkers of the prothrombotic state in abdominal obesity. Nutr. Hosp. 2014, 31, 1059–1066.
- Raoux, L.; Moszkowicz, D.; Vychnevskaia, K.; Poghosyan, T.; Beauchet, A.; Clauser, S.; Bretault, M.; Czernichow, S.; Carette, C.; Bouillot, J.-L. Effect of Bariatric Surgery-Induced Weight Loss on Platelet Count and Mean Platelet Volume: A 12-Month Follow-Up Study. Obes. Surg. 2017, 27, 387–393.
- Bielinski, S.J.; Berardi, C.; Decker, P.A.; Kirsch, P.S.; Larson, N.B.; Pankow, J.S.; Sale, M.; de Andrade, M.; Sicotte, H.; Tang, W.; et al. P-selectin and subclinical and clinical atherosclerosis: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2015, 240, 3–9.
- De Pergola, G.; Pannacciulli, N.; Coviello, M.; Scarangella, A.; Di Roma, P.; Caringella, M.; Venneri, M.T.; Quaranta, M.; Giorgino, R. sP-selectin plasma levels in obesity: Association with insulin resistance and related metabolic and prothrombotic factors. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 227–232.
- Audoly, L.P.; Rocca, B.; Fabre, J.E.; Koller, B.H.; Thomas, D.; Loeb, A.L.; Coffman, T.M.; FitzGerald, G.A. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation 2000, 101, 2833–2840.
- Horng, T.; Hotamisligil, G.S. Linking the inflammasome to obesity-related disease. Nat. Med. 2011, 17, 164–165.
- Murakami, T.; Horigome, H.; Tanaka, K.; Nakata, Y.; Ohkawara, K.; Katayama, Y.; Matsui, A. Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb. Res. 2007, 119, 45–53.
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; Ní Áinle, F.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles From Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk With Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7.
- Russo, I.; Del Mese, P.; Doronzo, G.; De Salve, A.; Secchi, M.; Trovati, M.; Anfossi, G. Platelet resistance to the antiaggregatory cyclic nucleotides in central obesity involves reduced phosphorylation of vasodilator-stimulated phosphoprotein. Clin. Chem. 2007, 53, 1053–1060.
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Doronzo, G.; De Salve, A.; Trovati, M. Impaired synthesis and action of antiaggregating cyclic nucleotides in platelets from obese subjects: Possible role in platelet hyperactivation in obesity. Eur. J. Clin. Investig. 2004, 34, 482–489.
- Russo, I.; Viretto, M.; Barale, C.; Mattiello, L.; Doronzo, G.; Pagliarino, A.; Cavalot, F.; Trovati, M.; Anfossi, G. High glucose inhibits the aspirin-induced activation of the nitric oxide/cGMP/cGMP-dependent protein kinase pathway and does not affect the aspirin-induced inhibition of thromboxane synthesis in human platelets. Diabetes 2012, 61, 2913–2921.
- Kawahara, Y.; Yamanishi, J.; Fukuzaki, H. Inhibitory action of guanosine 3′,5′-monophosphate on thrombin-induced calcium mobilization in human platelets. Thromb. Res. 1984, 33, 203–209.
- Resnick, L.M. Cellular ions in hypertension, insulin resistance, obesity, and diabetes: A unifying theme. J. Am. Soc. Nephrol. 1992, 3, S78–S85.
- Anfossi, G.; Mularoni, E.M.; Burzacca, S.; Ponziani, M.C.; Massucco, P.; Mattiello, L.; Cavalot, F.; Trovati, M. Platelet resistance to nitrates in obesity and obese NIDDM, and normal platelet sensitivity to both insulin and nitrates in lean NIDDM. Diabetes Care 1998, 21, 121–126.
- Hou, L.; Wang, Q.; Pan, B.; Li, R.; Li, Y.; He, J.; Qin, T.; Cao, L.; Zhang, N.; Cao, C.; et al. Exercise modalities for type 2 diabetes: A systematic review and network meta-analysis of randomized trials. Diabetes Metab. Res. Rev. 2023, 39, e3591.
- Khalafi, M.; Symonds, M.E.; Ghasemi, F.; Rosenkranz, S.K.; Rohani, H.; Sakhaei, M.H. The effects of exercise training on postprandial glycemia and insulinemia in adults with overweight or obesity and with cardiometabolic disorders: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2023, 201, 110741.
- Rauramaa, R.; Salonen, J.T.; Seppänen, K.; Salonen, R.; Venäläinen, J.M.; Ihanainen, M.; Rissanen, V. Inhibition of platelet aggregability by moderate-intensity physical exercise: A randomized clinical trial in overweight men. Circulation 1986, 74, 939–944.
- Keating, F.K.; Schneider, D.J.; Savage, P.D.; Bunn, J.Y.; Harvey-Berino, J.; Ludlow, M.; Toth, M.J.; Ades, P.A. Effect of exercise training and weight loss on platelet reactivity in overweight patients with coronary artery disease. J. Cardiopulm. Rehabil. Prev. 2013, 33, 371–377.
- Lamprecht, M.; Moussalli, H.; Ledinski, G.; Leschnik, B.; Schlagenhauf, A.; Koestenberger, M.; Polt, G.; Cvirn, G. Effects of a single bout of walking exercise on blood coagulation parameters in obese women. J. Appl. Physiol. 2013, 115, 57–63.
- Skouras, A.Z.; Antonakis-Karamintzas, D.; Tsantes, A.G.; Triantafyllou, A.; Papagiannis, G.; Tsolakis, C.; Koulouvaris, P. The Acute and Chronic Effects of Resistance and Aerobic Exercise in Hemostatic Balance: A Brief Review. Sports 2023, 11, 74.
- Rigamonti, A.E.; Bollati, V.; Pergoli, L.; Iodice, S.; De Col, A.; Tamini, S.; Cicolini, S.; Tringali, G.; De Micheli, R.; Cella, S.G.; et al. Effects of an acute bout of exercise on circulating extracellular vesicles: Tissue-, sex-, and BMI-related differences. Int. J. Obes. 2020, 44, 1108–1118.
- Dallak, M.; Bin-Jaliah, I.; Sakr, H.F.; Al-Ani, B.; Haidara, M.A. Swim exercise inhibits hemostatic abnormalities in a rat model of obesity and insulin resistance. Arch. Physiol. Biochem. 2019, 125, 79–84.
- Wen, J.; Huang, Y.; Lu, Y.; Yuan, H. Associations of non-high-density lipoprotein cholesterol, triglycerides and the total cholesterol/HDL-c ratio with arterial stiffness independent of low-density lipoprotein cholesterol in a Chinese population. Hypertens. Res. 2019, 42, 1223–1230.
- Lacoste, L.; Lam, J.Y.; Hung, J.; Letchacovski, G.; Solymoss, C.B.; Waters, D. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation 1995, 92, 3172–3177.
- Morotti, A.; Barale, C.; Melchionda, E.; Russo, I. Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell. Int. J. Mol. Sci. 2022, 23, 11446.
- Davì, G.; Romano, M.; Mezzetti, A.; Procopio, A.; Iacobelli, S.; Antidormi, T.; Bucciarelli, T.; Alessandrini, P.; Cuccurullo, F.; Bittolo Bon, G. Increased levels of soluble P-selectin in hypercholesterolemic patients. Circulation 1998, 97, 953–957.
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703.
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116.
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095.
- Betteridge, D.J.; Cooper, M.B.; Saggerson, E.D.; Prichard, B.N.; Tan, K.C.; Ling, E.; Barbera, G.; McCarthy, S.; Smith, C.C. Platelet function in patients with hypercholesterolaemia. Eur. J. Clin. Invest. 1994, 24 (Suppl. S1), 30–33.
- Combescure, C.; Fontana, P.; Mallouk, N.; Berdague, P.; Labruyere, C.; Barazer, I.; Gris, J.C.; Laporte, S.; Fabbro-Peray, P.; Reny, J.L.; et al. Clinical implications of clopidogrel non-response in cardiovascular patients: A systematic review and meta-analysis. J. Thromb. Haemost. 2010, 8, 923–933.
- Sofi, F.; Marcucci, R.; Gori, A.M.; Giusti, B.; Abbate, R.; Gensini, G.F. Clopidogrel non-responsiveness and risk of cardiovascular morbidity. An updated meta-analysis. Thromb. Haemost. 2010, 103, 841–848.
- Stone, G.W.; Witzenbichler, B.; Weisz, G.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; Cox, D.A.; Duffy, P.L.; Mazzaferri, E.; et al. Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): A prospective multicentre registry study. Lancet 2013, 382, 614–623.
- Valiyaveettil, M.; Podrez, E.A. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J. Thromb. Haemost. 2009, 7 (Suppl. S1), 218–221.
- Ma, Y.; Ashraf, M.Z.; Podrez, E.A. Scavenger receptor BI modulates platelet reactivity and thrombosis in dyslipidemia. Blood 2010, 116, 1932–1941.
- Akkerman, J.W.N. From low-density lipoprotein to platelet activation. Int. J. Biochem. Cell Biol. 2008, 40, 2374–2378.
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell Longev. 2019, 2019, 8267234.
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 2014, 129, 1598–1609.
- Pawlowska, Z.; Swiatkowska, M.; Krzeslowska, J.; Pawlicki, L.; Cierniewski, C.S. Increased platelet-fibrinogen interaction in patients with hypercholesterolemia and hypertriglyceridemia. Atherosclerosis 1993, 103, 13–20.
- Relou, I.A.M.; Hackeng, C.M.; Akkerman, J.-W.N.; Malle, E. Low-density lipoprotein and its effect on human blood platelets. Cell Mol. Life Sci. 2003, 60, 961–971.
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 2005, 26, 33–65.
- Barale, C.; Frascaroli, C.; Cavalot, F.; Russo, I. Hypercholesterolemia impairs the Glucagon-like peptide 1 action on platelets: Effects of a lipid-lowering treatment with simvastatin. Thromb. Res. 2019, 180, 74–85.
- Willoughby, S.R.; Stewart, S.; Holmes, A.S.; Chirkov, Y.Y.; Horowitz, J.D. Platelet nitric oxide responsiveness: A novel prognostic marker in acute coronary syndromes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2661–2666.
- Riba, R.; Nicolaou, A.; Troxler, M.; Homer-Vaniasinkam, S.; Naseem, K.M. Altered platelet reactivity in peripheral vascular disease complicated with elevated plasma homocysteine levels. Atherosclerosis 2004, 175, 69–75.
- Barale, C.; Buracco, S.; Cavalot, F.; Frascaroli, C.; Guerrasio, A.; Russo, I. Glucagon-like peptide 1-related peptides increase nitric oxide effects to reduce platelet activation. Thromb. Haemost. 2017, 117, 1115–1128.
- Alber, H.F.; Frick, M.; Suessenbacher, A.; Doerler, J.; Schirmer, M.; Stocker, E.-M.; Dichtl, W.; Pachinger, O.; Weidinger, F. Effect of atorvastatin on circulating proinflammatory T-lymphocyte subsets and soluble CD40 ligand in patients with stable coronary artery disease--a randomized, placebo-controlled study. Am. Heart J. 2006, 151, 139.
- Pignatelli, P.; Carnevale, R.; Cangemi, R.; Loffredo, L.; Sanguigni, V.; Stefanutti, C.; Basili, S.; Violi, F. Atorvastatin inhibits gp91phox circulating levels in patients with hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 360–367.
- Bea, F.; Blessing, E.; Shelley, M.I.; Shultz, J.M.; Rosenfeld, M.E. Simvastatin inhibits expression of tissue factor in advanced atherosclerotic lesions of apolipoprotein E deficient mice independently of lipid lowering: Potential role of simvastatin-mediated inhibition of Egr-1 expression and activation. Atherosclerosis 2003, 167, 187–194.
- Tannous, M.; Cheung, R.; Vignini, A.; Mutus, B. Atorvastatin increases ecNOS levels in human platelets of hyperlipidemic subjects. Thromb. Haemost. 1999, 82, 1390–1394.
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Loffredo, L.; Carnevale, R.; Sorge, R.; Violi, F. Oxidative stress-mediated platelet CD40 ligand upregulation in patients with hypercholesterolemia: Effect of atorvastatin. J. Thromb. Haemost. 2007, 5, 1170–1178.
- Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’Erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193.
- Pedersen, B.K.; Saltin, B. Evidence for prescribing exercise as therapy in chronic disease. Scand. J. Med. Sci. Sports 2006, 16 (Suppl. S1), 3–63.
- Aadahl, M.; Kjaer, M.; Jørgensen, T. Associations between overall physical activity level and cardiovascular risk factors in an adult population. Eur. J. Epidemiol. 2007, 22, 369–378.
- Mann, S.; Beedie, C.; Jimenez, A. Differential effects of aerobic exercise, resistance training and combined exercise modalities on cholesterol and the lipid profile: Review, synthesis and recommendations. Sports Med. 2014, 44, 211–221.
- Alvarez-Jimenez, L.; Moreno-Cabañas, A.; Ramirez-Jimenez, M.; Morales-Palomo, F.; Ortega, J.F.; Mora-Rodriguez, R. Effectiveness of statins vs. exercise on reducing postprandial hypertriglyceridemia in dyslipidemic population: A systematic review and network meta-analysis. J. Sport. Health Sci. 2022, 11, 567–577.
- Pearson, R.C.; Cogan, B.; Garcia, S.A.; Jenkins, N.T. Effect of Prior Exercise on Postprandial Lipemia: An Updated Meta-Analysis and Systematic Review. Int. J. Sport. Nutr. Exerc. Metab. 2022, 32, 501–518.
- Katzman, P.L.; Bose, R.; Henry, S.; McLean, D.L.; Walker, S.; Fyfe, C.; Perry, Y.; Mymin, D.; Bolli, P. Serum lipid profile determines platelet reactivity to native and modified LDL-cholesterol in humans. Thromb. Haemost. 1994, 71, 627–632.
- Weidtmann, A.; Scheithe, R.; Hrboticky, N.; Pietsch, A.; Lorenz, R.; Siess, W. Mildly oxidized LDL induces platelet aggregation through activation of phospholipase A2. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1131–1138.
- Hsu, H.C.; Lee, Y.T.; Chen, M.F. Exercise shifts the platelet aggregation modulatory role from native to mildly oxidized low-density lipoprotein. Med. Sci. Sports Exerc. 2000, 32, 933–939.
- Wang, J.-S.; Chow, S.-E. Effects of exercise training and detraining on oxidized low-density lipoprotein-potentiated platelet function in men. Arch. Phys. Med. Rehabil. 2004, 85, 1531–1537.
- Su, X.; Yu, X.; Chen, R.; Bian, W. Swimming improves platelet dysfunction in mice fed with a high-fat diet. Arch. Physiol. Biochem. 2023, 129, 198–203.
- Vazzana, N.; Ganci, A.; Cefalù, A.B.; Lattanzio, S.; Noto, D.; Santoro, N.; Saggini, R.; Puccetti, L.; Averna, M.; Davì, G. Enhanced lipid peroxidation and platelet activation as potential contributors to increased cardiovascular risk in the low-HDL phenotype. J. Am. Heart Assoc. 2013, 2, e000063.
- Sonmez, O.; Sonmez, M. Role of platelets in immune system and inflammation. Porto Biomed. J. 2017, 2, 311–314.
- Carr, M.E. Diabetes mellitus: A hypercoagulable state. J. Diabetes Complicat. 2001, 15, 44–54.
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121.
- Ghoshal, K.; Bhattacharyya, M. Overview of platelet physiology: Its hemostatic and nonhemostatic role in disease pathogenesis. Sci. World J. 2014, 2014, 781857.
- Bosco, O.; Vizio, B.; Gruden, G.; Schiavello, M.; Lorenzati, B.; Cavallo-Perin, P.; Russo, I.; Montrucchio, G.; Lupia, E. Thrombopoietin Contributes to Enhanced Platelet Activation in Patients with Type 1 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 7032.
- Ferreiro, J.L.; Gómez-Hospital, J.A.; Angiolillo, D.J. Platelet abnormalities in diabetes mellitus. Diabetes Vasc. Dis. Res. 2010, 7, 251–259.
- Santilli, F.; Simeone, P.G.; Guagnano, M.T.; Leo, M.; Maccarone, M.T.; Di Castelnuovo, A.; Sborgia, C.; Bonadonna, R.C.; Angelucci, E.; Federico, V.; et al. Effects of Liraglutide on Weight Loss, Fat Distribution, and β-Cell Function in Obese Subjects With Prediabetes or Early Type 2 Diabetes. Diabetes Care 2017, 40, 1556–1564.
- Santilli, F.; Formoso, G.; Sbraccia, P.; Averna, M.; Miccoli, R.; Di Fulvio, P.; Ganci, A.; Pulizzi, N.; Lattanzio, S.; Ciabattoni, G.; et al. Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. J. Thromb. Haemost. 2010, 8, 828–837.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853.
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559.
- ADVANCE Collaborative Group; Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572.
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139.
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322.
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Abdul-Ghani, M.; DeFronzo, R.A.; Del Prato, S.; Chilton, R.; Singh, R.; Ryder, R.E.J. Cardiovascular Disease and Type 2 Diabetes: Has the Dawn of a New Era Arrived? Diabetes Care 2017, 40, 813–820.
- Way, K.L.; Hackett, D.A.; Baker, M.K.; Johnson, N.A. The Effect of Regular Exercise on Insulin Sensitivity in Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Metab. J. 2016, 40, 253–271.
- Marwick, T.H.; Hordern, M.D.; Miller, T.; Chyun, D.A.; Bertoni, A.G.; Blumenthal, R.S.; Philippides, G.; Rocchini, A.; Council on Clinical Cardiology, American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee; Council on Cardiovascular Disease in the Young; et al. Exercise training for type 2 diabetes mellitus: Impact on cardiovascular risk: A scientific statement from the American Heart Association. Circulation 2009, 119, 3244–3262.
- Akbarinia, A.; Kargarfard, M.; Naderi, M. Aerobic training improves platelet function in type 2 diabetic patients: Role of microRNA-130a and GPIIb. Acta Diabetol. 2018, 55, 893–899.
- Taghizadeh, M.; Kargarfard, M.; Braune, S.; Jung, F.; Naderi, M. Long-term aerobic exercise training in type two diabetic patients alters the expression of miRNA-223 and its corresponding target, the P2RY12 receptor, attenuating platelet function. Clin. Hemorheol. Microcirc. 2022, 80, 107–116.
- Taghizadeh, M.; Ahmadizad, S.; Naderi, M. Effects of endurance training on hsa-miR-223, P2RY12 receptor expression and platelet function in type 2 diabetic patients. Clin. Hemorheol. Microcirc. 2018, 68, 391–399.
- Bratseth, V.; Chiva-Blanch, G.; Byrkjeland, R.; Solheim, S.; Arnesen, H.; Seljeflot, I. Elevated levels of circulating microvesicles in coronary artery disease patients with type 2 diabetes and albuminuria: Effects of exercise training. Diabetes Vasc. Dis. Res. 2019, 16, 431–439.
- Çakır, H.; Kaymaz, C.; Tanboga, İ.H.; Çakır, H.; Tokgöz, H.C.; Hakgör, A.; Akbal, Ö.Y.; Er, F.; Topal, D.; Mutluer, F.O.; et al. Increased exercise-related platelet activation assessed by impedance aggregometry in diabetic patients despite aspirin therapy. J. Thromb. Thrombolysis 2019, 47, 396–402.
- Whyte, J.J.; Laughlin, M.H. The effects of acute and chronic exercise on the vasculature. Acta Physiol. 2010, 199, 441–450.
- Francois, M.E.; Myette-Cote, E.; Bammert, T.D.; Durrer, C.; Neudorf, H.; DeSouza, C.A.; Little, J.P. Carbohydrate restriction with postmeal walking effectively mitigates postprandial hyperglycemia and improves endothelial function in type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H105–H113.
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354.
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532.
- Ceriello, A.; Kumar, S.; Piconi, L.; Esposito, K.; Giugliano, D. Simultaneous control of hyperglycemia and oxidative stress normalizes endothelial function in type 1 diabetes. Diabetes Care 2007, 30, 649–654.
- Scheinowitz, M.; Pakala, R.; Ben-Dor, I.; Lemesle, G.; Torguson, R.; Pichard, A.D.; Lindsay, J.; Waksman, R. Platelet reactivity in diabetic patients subjected to acute exercise stress test. Cardiovasc. Revasc. Med. 2011, 12, 20–24.
- Fini, E.M.; Salimian, M.; Ahmadizad, S. Responses of platelet CD markers and indices to resistance exercise with and without blood flow restriction in patients with type 2 diabetes. Clin. Hemorheol. Microcirc. 2022, 80, 281–289.