Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Viviana Moresi.
Histone deacetylases (HDACs) are enzymes that regulate the deacetylation of numerous histone and non-histone proteins, thereby affecting a wide range of cellular processes. Deregulation of HDAC expression or activity is often associated with several pathologies, suggesting potential for targeting these enzymes for therapeutic purposes.
- histone deacetylase
- muscular dystrophies
- Duchenne Muscular Dystrophy
1. Overview of the Histone Deacetylases and Their Role in Striated Muscles
Transcriptional regulation in eukaryotes is strongly influenced by post-translational modifications (PTMs) of histones, the core proteins of chromatin, such as phosphorylation, methylation, and acetylation. Histone acetylation is probably the most well-characterized of these modifications, with hyperacetylation leading to an increase in gene expression, due to the relaxation of chromatin structure, while hypoacetylation has the opposite effect. The latter is mediated by histone deacetylases (HDACs) [1]. By doing this, HDACs influence the delicate balance between euchromatin and heterochromatin, thereby widely affecting gene expression in a prolonged fashion [2]. Therefore, the balance between the levels of histone deacetylation and acetylation plays a key role in the modulation of gene transcription and governs numerous developmental processes, being involved in the regulation of various genes associated with signal transduction, cell growth, and cell death, as well as disease states, including fluid and electrolyte disorders or cancers [3,4][3][4]. In addition, HDACs deacetylate non-histone proteins, such as p53 [5,6][5][6] as one of the first identified HDAC targets, thus regulating their activity.
The numerous HDACs have a wide range of expression and function in multiple cell types and tissues. In spite of the lack of complete knowledge of their roles, a global inhibition of deacetylase activity in the human body has been proposed as a therapeutical approach for various disease states, including muscle dystrophy. Before approaching this issue, it is therefore important to provide an overview of the HDAC family, focusing on the different roles HDACs play in striated muscle. According to their sequence similarities with yeast orthologs and the use of either Zn2+ or NAD+ as cofactors [3[3][7][8],7,8], 18 human HDACs have been identified and grouped into four classes.
Class I HDACs shows similarity to the yeast deacetylase Rpd3p enzyme and include HDAC1, 2, 3 and 8. They are Zn2+-dependent, ubiquitously expressed enzymes, which are localized prevalently in the nucleus, playing a key role in the lysine deacetylation of N-terminal histone tails [9]. They are essential regulators of gene expression, being recruited to specific chromatin loci as a part of multi-protein complexes that control the acetylation state of histones and other chromatin-associated factors [10], resulting in chromatin condensation and transcriptional silencing [11]. The best-studied complexes include the NuRD, Sin3 and CoREST complexes, which contain HDAC1/2, and the SMRT/NCoR complex, which contains HDAC3 [12,13,14][12][13][14].
Thanks to tissue-specific knock-out (KO) mouse models, it has been established that HDAC1 and 2 often play redundant functions in the development or homeostasis maintenance of numerous tissues and cell types. In the heart, HDAC1 and 2 repress genes encoding contractile proteins and calcium channels [15], while, in skeletal muscle, they control autophagic flux and muscle metabolism [16].
HDAC3 is required for normal mouse development and tissue-specific functions by epigenetically controlling metabolism and circadian rhythms [17]. Cell type- or tissue-specific deletion reveal a role of HDAC3 in cardiac development and cardiomyocyte metabolism, since its absence leads to severe underdevelopment of the ventricular walls and to ventricular septal defects [18,19,20][18][19][20]. Of note, some of these functions in cardiac development are independent of its deacetylase activity; rather HDAC3 regulates gene transcription by recruiting other epigenetic factors to the NCOR complex [19] or by tethering peripheral heterochromatin to the nuclear lamina [18]. As a demonstration of the importance of HDAC3 in the whole-body metabolism, the deletion of Hdac3 in skeletal muscle causes severe systemic and skeletal muscle-specific insulin resistance, impaired insulin and glucose tolerance, and diminished glucose uptake into skeletal muscle, overall impacting on muscle performance [21].
HDAC8 controls processes different from the other class I HDAC members and has a multifaceted role in human pathophysiology [22]. However, to date, no specific role in striated muscle has been reported.
Class II HDACs are similar to the yeast Hda1 deacetylase enzyme. This class is further subdivided into class IIa (HDACs 4, 5, 7 and 9) and class IIb (HDACs 6 and 10). While class IIa HDACs localize both in the nucleus and in the cytoplasm, class IIb HDACs are primarily in the cytoplasm and contain two catalytic sites. Class IIa HDACs are characterized by an extended N-terminal domain, containing conserved serine (Ser) residues, which are subjected to phosphorylation by several kinases, such as CaMK or SIK [23[23][24][25],24,25], facilitating HDAC nuclear export. Moreover, because of a Tyr-to-His mutation in their catalytic pocket, class IIa HDACs possess very low deacetylase activity compared to class I and class IIb HDACs [26]. This finding implies that class IIa HDACs regulate gene transcription via acting as scaffold proteins to recruit class I HDACs to specific genes, or as tethering proteins to anchor chromatin regions, or via sterically block transcription factor activity.
Among the members of class IIa, HDAC4 plays crucial functions in striated muscles. Increased expression of HDAC4 has been detected in skeletal muscle in different diseases, such as Duchenne Muscular Dystrophy (DMD) [27] and Amyotrophic Lateral Sclerosis (ALS) [28,29][28][29]: importantly, the observations in pre-clinical models have been validated in patients. Despite binding and repressing the activity of two major myogenic factors, i.e., MEF2 [30] and SRF [31], mice harboring a skeletal-muscle specific deletion of Hdac4 are viable and do not display obvious defects in skeletal muscle [32]. While class IIa HDACs play redundant roles in the establishment of the metabolic pattern of skeletal muscle fibers, by repressing MEF2 [33], HDAC4 per se is necessary and sufficient to mediate responses upon different stimuli in skeletal muscle. For instance, deletion of Hdac4 in differentiating skeletal muscle cells via myogenin:Cre recombinase, hampers muscle regeneration, due to the release of soluble factors that inhibit muscle precursor cell differentiation [34]; if the deletion of Hdac4 occurs earlier in the myogenic cells, such as in Pax7+ cells, it compromises muscle stem cell (MuSC) proliferation and differentiation [35]. Together with HDAC5, HDAC4 connects neural activity to skeletal muscle transcription upon denervation, via both epigenetic regulation of gene expression [36,37,38][36][37][38] and by modulating nuclear and cytoplasmic non-histone protein acetylation [39[39][40],40], thereby mediating neurogenic muscle atrophy. Interestingly, deletion of Hdac4 in skeletal muscle is protective in experimental models of neurogenic muscle atrophy in the early phases after the surgical procedure [37]; however, it resulted detrimental effects following long-term denervation, causing muscle degeneration due to the impairment in the activation of multiple signaling, including the oxidative stress response, the ubiquitin-proteasome system and the autophagic pathway [32]. Consistently, deletion of Hdac4 in skeletal muscle in a mouse model of ALS worsened pathological features, advancing and exacerbating skeletal muscle atrophy and denervation by modulating several biological processes and gene networks [29]. Similar to ALS, HDAC4 expression is upregulated in DMD skeletal muscles [27], and, consistently, deletion of Hdac4 results detrimental in both disease states. Indeed, mdx mice with a skeletal muscle-specific deletion of HDAC4 show increased muscle damage and hampered muscle regeneration, overall leading to decreased muscle function. HDAC4 prevalently localizes in the cytoplasm of dystrophic muscles, where it mediates activation of the membrane repair mechanism, likely through a deacetylase-independent activity, thereby affecting muscle necrosis, satellite cell survival and myogenic capacity [27]. Overall, these studies suggest that skeletal muscle up-regulates HDAC4 expression upon stress as a response to a disease state. In the heart, the N-terminal proteolytically derived fragment of HDAC4 finely regulates lipid metabolism and glucose handling through MEF2-dependent gene expression, ultimately protecting from heart failure [41,42][41][42].
HDAC5 acts as a negative epigenetic regulator of IL-6 synthesis and release in skeletal muscle, and Hdac5 global KO mice show improved systemic glucose tolerance in response to exercise [43]. Of note, a non-deacetylase-dependent regulatory role of HDAC5 has been reported in cardiac cells. By using Hdac5 global KO mice, it has been illustrated that HDAC5 is required for the interaction of the class I HDAC/Sin3 co-repressor complex with the Nkx2.5 and YY1 transcription factors and the consequent recruitment of the complex to promoter regions of either the Ncx1 or Bnp gene, which are important for cardiac hypertrophy [44].
HDAC9 is highly expressed in cardiac muscle even though it does not affect heart development. Nonetheless, mutant mice lacking Hdac9 are sensitized to hypertrophic signals and exhibit stress-dependent cardiomegaly, suggesting that HDAC9 is a negative regulator of cardiomyocyte hypertrophy [45,46][45][46]. HDAC9 mutant mice showed an increase in slow fibers suggesting that its deletion results in enhanced slow-fiber gene expression [33].
Among class IIb HDACs, HDAC6 has been found associated with the class III deacetylase SIRT2 [47]. This complex interacts with poly-ubiquitin and poly-ubiquitinated proteins [48], and with tubulin and microtubules in the cytoplasm [49]. In particular, it has been observed that HDAC6 localizes at NMJs and its deletion protects against microtubule disorganization, markedly influencing NMJ structure [50]. Moreover, HDAC6 expression is upregulated during muscle atrophy, where it interacts with the E3-ubiquitin ligase MAFbx, participating to its activation; consistently, HDAC6 inactivation protects against muscle wasting in mice [51]. In the heart, HDAC6 was recently found to regulate myofibril stiffness and diastolic function [52].
HDAC10 mainly acts as polyamine deacetylase instead of lysine deacetylase [53], but its targets and functions in striated muscle are poorly characterized.
Class III HDACs show similarity to the yeast Sir2. In humans, the family consists of seven members, named sirtuins (SIRT1-7), whose activity depend on NAD+ [54]. Sirt1 KO mice are sterile, smaller and present abnormalities in heart morphogenesis, due to p53 hyperacetylation and p53-dependent apoptosis [55]. Moreover, SIRT1 plays an essential role in the maintenance of mitochondrial integrity by modulating the MEF2 transcription factors in the heart [56]. Similarly to the yeast Sir2, SIRT1 exerts longevity effects against aging-associated pathologies, such as neurodegeneration, metabolic dysfunction [57], and cardiovascular diseases [58]. Consistently, SIRT1 levels decrease with age [59], promoting senescence. In skeletal muscle, SIRT1 inhibits FoxO1 and FoxO3 activity upon fasting, thereby protecting muscle from atrophy while promoting growth [60]. Importantly, SIRT1 is a sensor of energy metabolism, being triggered by AMPK, and deacetylates, thus activating, peroxisome proliferator activated receptor gamma coactivator 1α (PGC-1α) [61].
2. Histone Deacetylases in Muscular Dystrophies
Several HDAC isoforms have been implicated in skeletal muscle remodeling, both in physiological and pathological conditions [95,96][62][63]. Ample work revealed that HDACs exert pivotal roles in regulating fiber type specification [96][63], muscle fiber size and innervation [29,37[29][37][64],97], metabolic fuel switching [16[16][65][66],98,99], muscle development [100][67], insulin sensitivity and exercise capacity [69[68][69][70],101,102], thus contributing to the maintenance of skeletal muscle homeostasis. The evidence of a wide variety of HDAC functions in skeletal muscle led to an increasing interest to clarify their roles in skeletal muscle disorders [29[29][63],96], including muscular dystrophies (MDs). MDs consist of a heterogeneous group of genetic disorders characterized by progressive weakness and degeneration of skeletal muscles resulting in impaired muscle function [103][71]. Traditionally classified by a patient’s clinical presentation, muscle group involvement, mode of inheritance, age of onset and overall disease progression, MDs have been linked to a variety of distinct single-gene mutations [104][72]. So far, molecular genetic mapping techniques have shown that MDs are caused by numerous mutations in several genes encoding structural and functional muscle proteins, resulting in degeneration or dysfunction of skeletal muscle [104][72]. The most severe and the most common adult form of MD is Duchenne Muscular Dystrophy (DMD), which affects 1 in 3500–6000 live male births, and is caused by the lack of functional dystrophin protein due to mutations in the dystrophin gene (DMD) [105][73]. The structural role of dystrophin is closely related to its centrality in assembling the sarcolemmal Dystrophin-Associated Protein Complex (DAPC), which provides the molecular link between the cytoskeleton and the extracellular matrix of skeletal myofibers [106,107][74][75]. Lack of dystrophin results in mechanical instability causing myofibers rupture during contraction. Moreover, being connected with multiple proteins, dystrophin modulates several signal transduction pathways, including Ca2+ entry, nitric oxide (NO), and reactive oxygen species (ROS) production [108,109,110][76][77][78]. The mdx mouse, harboring a nonsense point mutation in the exon 23 that aborts the full-length dystrophin expression, is the most widely used animal model for DMD research [111][79]. Despite the loss of dystrophin, mdx mice show minimal clinical features of the disease, if compared with DMD patients, probably due to compensatory mechanisms. The latter include muscle regeneration, which is more efficient in mdx mice compared to DMD patients, in part due to differences in telomere shortening and muscle stem cell regenerative capacity [112][80]. Among compensatory mechanisms triggered by the absence of dystrophin, the upregulation of utrophin has been reported in both DMD and mdx myofibers [113][81]. Utrophin is a structural and functional autosomal paralogue of dystrophin, normally located at the neuromuscular and myotendinous junctions in adult skeletal muscle in physiological condition [114][82], but enriched at the sarcolemma in dystrophic myofibers where it acts to preserving muscle function and mitigating necrosis [113][81]. Importantly, while the exogenous expression of utrophin attenuated the mdx dystrophic phenotype, its deletion in mdx mice worsened the pathology, thus confirming that utrophin protective functions in DMD [115,116,117][83][84][85]. In addition to dystrophin, another important member of the DAPC is the sarcoglycan complex, which is composed of four sarcoglycan (SG) proteins, α−, β−, δ−, and γ-SG, playing a key role to protect striated muscle membranes against contraction-induced damage [118,119][86][87]. Mutations in one of the four sarcoglycan genes (SGCA) cause a different form of autosomal recessive sarcoglycanopathies [120[88][89],121], a subgroup of Limb Girdle Muscular Dystrophies (LGMDs). Sarcoglycanopathies are more frequently found among the most severe forms of MDs, and the clinical phenotype closely resembles that of DMD, with onset during childhood [122,123][90][91]. The role of HDACs in MDs is not yet fully identified; indeed, most of our knowledge derives from studies with HDAC inhibitors in dystrophic contexts (discussed below). However, several studies revealed the deregulation of HDAC expression or activity in dystrophic muscles (Figure 1).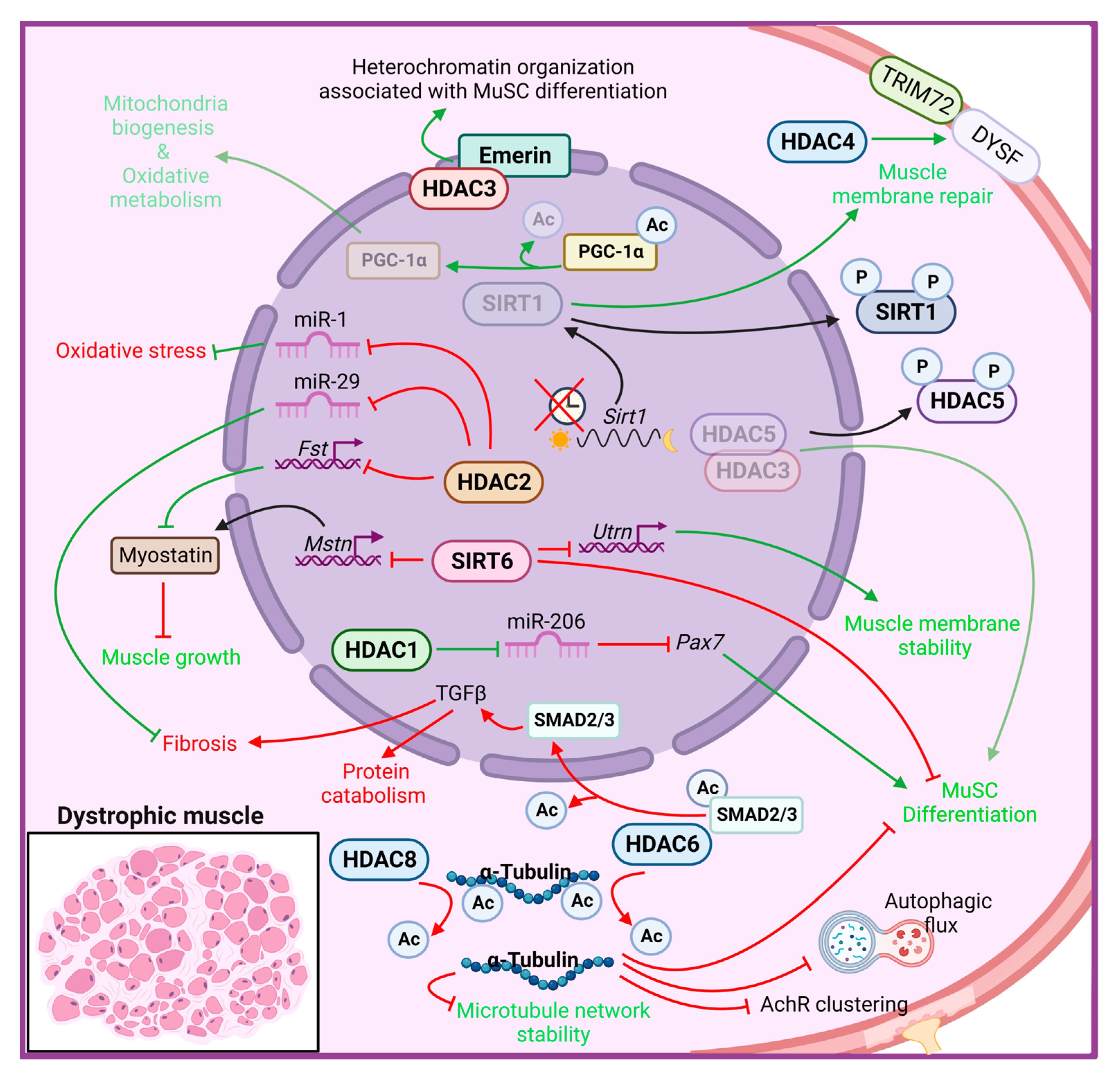
Figure 1. Histone deacetylase functions in muscle dystrophy condition. The cellular responses that promote MD progression are indicated in red, while in green those that counteract MD pathological features. AChR: acetylcholine receptor; Ac: acetyl group; Fst: follistatin; Mstn: myostatin; Utrn: utrophin; HDAC: histone deacetylase; SIRT: sirtuin; DYSF: Dysferlin; PGC-1α: peroxisome proliferator-activated receptor, gamma, coactivator 1 alpha; TGF-β: transforming growth factor beta.
3. Targeting Histone Deacetylases in Muscular Dystrophies
Epigenetic mechanisms controlling transcriptional programs in tissue progenitors are becoming a critical area of interest in medicine. Indeed, current studies are focused on manipulating chromatin targets of individual signaling pathways to provide novel regenerative strategies based on epigenetic drug administration. Numerous studies have highlighted the fundamental role of HATs and HDACs in regulating muscle gene transcription and therefore, muscle development and differentiation. Moreover, cumulative in vitro and in vivo evidence in the last years has underscored the link between HDAC deregulation and the pathogenesis of several MDs, in particular of the most severe one, the DMD [162,163,164][133][134][135]. In this context, HDACi have been shown to act in a selective way, potentiating myogenesis through the hyperacetylation of genes regulated during development and resolving their epigenetic bivalency, a characteristic signature that identifies genes poised for transcription that typically are enriched in embryonic stem cells or pluripotent cells [165][136]. Starting from this evidence, by inhibiting HDACs and reestablishing the epigenetic events necessary to activate adult stem cells, it represents one of the most powerful approaches to restoring the downstream networks of muscle regeneration and muscle homeostasis, leading to increased functional and morphological recovery of dystrophic muscles. At first, focusing on the HDACi activity on skeletal muscle cells in vitro, it was observed that the pharmacological treatment targets myogenic differentiation [97,166][64][137]. Indeed, treatment of wild-type myoblasts with pan-HDACi, such as Trichostatin A (TSA), Valproic acid (VPA), or Sodium Butyrate (PhB), increases their differentiation potential and fusion capacity, due to different mechanisms: (i) the upregulation of MyoD acetylation; (ii) the modulation of histone acetylation at specific gene promoters and (iii) the increase of the expression of the pro-myogenic protein follistatin [97,166][64][137]. Several years ago, a link between dystrophin loss and HDAC activity was demonstrated [124,125][92][93]. In mdx whole muscles and primary myoblasts, an increase in global HDAC activity and HDAC2 expression was observed in association with a reduction in follistatin expression. Inhibition of HDAC2, by using the class I HDAC inhibitor MS-275 or siRNA, restores the level of global HDAC activity similar to healthy control muscles, leading to morphological and functional benefits in dystrophic muscles [124][92]. In more recent studies, increased activity of class I, class IIa and class I/IIb HDACs in muscles of 1.5-month-old mdx mice [27] and in Fibro-Adipogenic Progenitors (FAPs) isolated from 1.5 month- and 12 month-old mdx mice has been reported [167][138], further suggesting the involvement of HDACs in the pathogenesis of DMD. Next-generation sequencing studies have focused on the fine regulation of myogenesis by HDACi, paying attention to the epigenetic players that create changes in the epigenome, opening new therapeutic options in muscle diseases. It emerged that most of the beneficial effects of the HDACi on dystrophic muscles arise from their ability to selectively activate a microRNA-SWI/SNF-based epigenetic network in FAPs, a specific population of mesenchymal cells resident in muscle interstitium [168,169][139][140]. FAPs are a muscle cell population that, while in regenerating conditions support MuSCs differentiation, in pathological conditions, such as DMD, contribute to the progression of the disease, affecting fibrotic and fat deposition, decreasing muscle contractility, and altering metabolism [170,171,172][141][142][143]. Intriguingly, pan-HDACi manipulate cell fate determination that redirects the lineage commitment of FAPs from a fibro-adipogenic toward a myogenic one [169][140]. In the context of MDs, it is worth mentioning the sirtuins, which are class III histone/protein deacetylases, are able to modulate several important physiological mechanisms such as inflammation, apoptosis, glucose homeostasis, life span, and neuroprotection. Acting pharmacologically on these enzymes permits modification of the acetylation state of several intracellular messengers, thereby regulating downstream mechanisms. This approach likely has strong therapeutic potential for many human diseases such as metabolic disorders, and degenerative diseases such as MDs. As described above, the most studied of the sirtuins is SIRT1, which is expressed in many tissues, including skeletal muscle and heart, where it deacetylates and activates PGC-1α, a key modulator of muscle metabolism. The activated form of PGC-1α controls mitochondrial biogenesis and homeostasis, and therefore SIRT1 modulation was seen to be associated with muscle pathologies. It is now well known that PGC-1α overexpression in dystrophic mdx mice leads to milder signs of pathology and an improved function both in normal condition and after intense physical exercise [61,145][61][113]. Other mechanistic roles are attributed to SIRT1 modulation, supporting the beneficial effects on muscle pathologies. It has been described for example that SIRT1 stimulates and restores autophagy in muscle tissue through the deacetylation of autophagy components, including Atg5, Atg7, and Atg8, and activating FoxO3a a transcription factor that regulates autophagy in skeletal muscle [173,174][144][145]. Moreover, SIRT1 may modulate the activity of SMAD transcription factors, key TGF-β signaling components that are involved in myofibroblast differentiation. The activity of SMAD is regulated by lysine acetylation/deacetylation, which plays a critical role in tissue fibrosis [175][146]. All these data generated in vitro on cells (Figure 2), together with the in vivo evidence of deregulated activity of HDACs in MDs, have provided the rationale for using pan-HDACi and modulators of sirtuins in preclinical studies, with the aim of assessing the ability of these classes of compounds to improve muscle regeneration and counteract muscle degeneration in models of MD.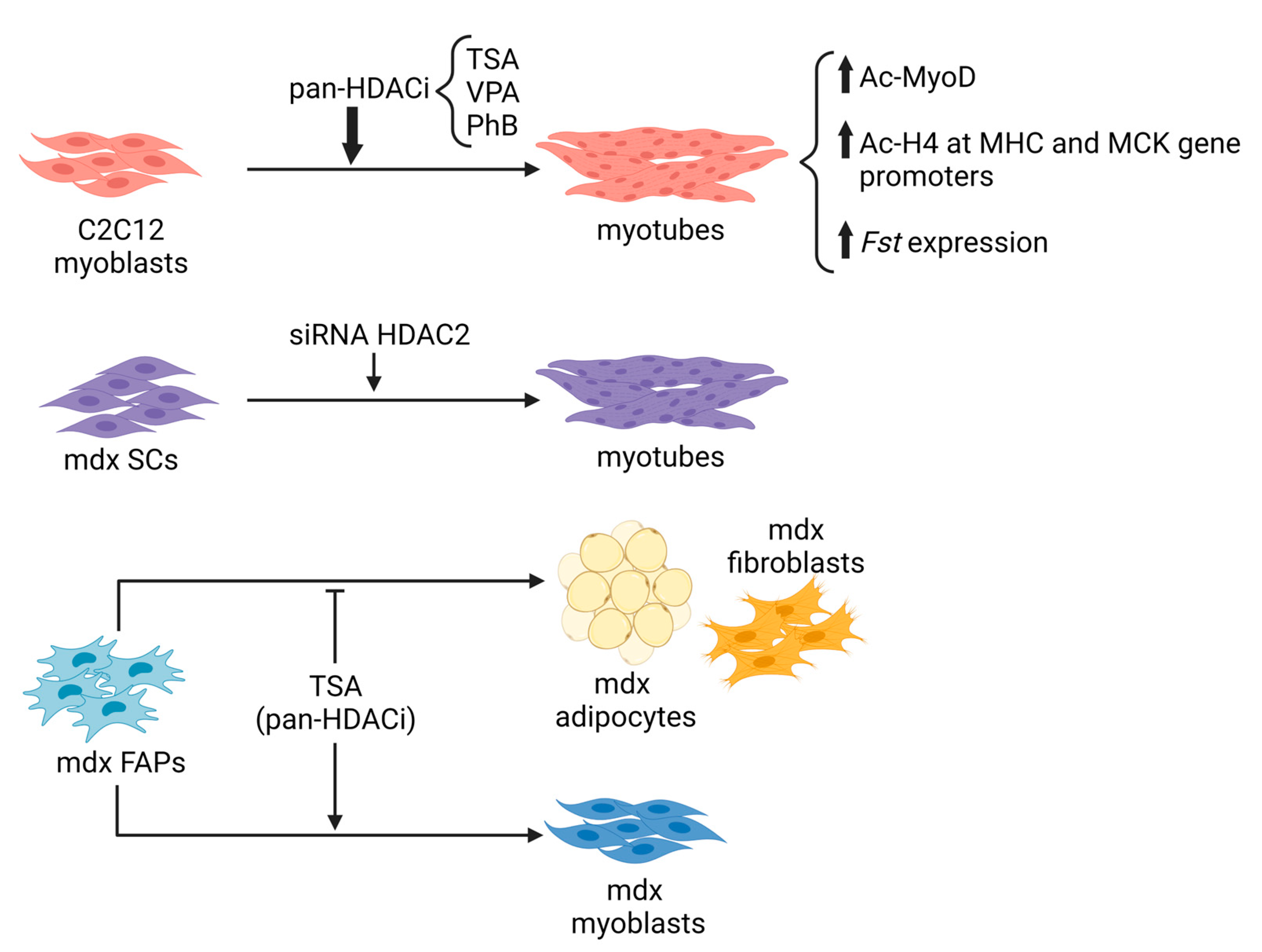
Figure 2. In vitro evidence of inhibiting HDACs on myoblast or FAP lineage progression. TSA: Trichostatin A; VPA: Valproic acid; PhB: Sodium Butyrate; Fst: follistatin; SCs: satellite cells; FAPs: Fibroadipogenic progenitors.
References
- De Ruijter, A.J.M.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B.P. Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem. J. 2003, 370, 737–749.
- Aygün, O.; Mehta, S.; Grewal, S.I.S. HDAC-Mediated Suppression of Histone Turnover Promotes Epigenetic Stability of Heterochromatin. Nat. Struct. Mol. Biol. 2013, 20, 547–554.
- Park, S.Y.; Kim, J.S. A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp. Mol. Med. 2020, 52, 204–212.
- Hyndman, K.A.; Knepper, M.A. Dynamic Regulation of Lysine Acetylation: The Balance between Acetyltransferase and Deacetylase Activities. Am. J. Physiol. Ren. Physiol. 2017, 313, F842–F846.
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174.
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-Mediated Deacetylation of P53 Is Required for Its Degradation. EMBO J. 2002, 21, 6236–6245.
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The Application of Histone Deacetylases Inhibitors in Glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138.
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and Characterization of a Novel Human Class I Histone Deacetylase That Functions as a Transcription Repressor. J. Biol. Chem. 2000, 275, 15254–15264.
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC Histone Deacetylase Complex Is Essential for Embryonic Development and Has a Unique Multivalent Structure. Nat. Commun. 2020, 11, 3252.
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate. Nature 2012, 481, 335–340.
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a Novel Complex with Both ATP-Dependent Chromatin-Remodeling and Histone Deacetylase Activities. Mol. Cell 1998, 2, 851–861.
- Laherty, C.D.; Yang, W.M.; Jian-Min, S.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone Deacetylases Associated with the MSin3 Corepressor Mediate Mad Transcriptional Repression. Cell 1997, 89, 349–356.
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, J.A.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural Basis for the Assembly of the SMRT/NCoR Core Transcriptional Repression Machinery. Nat. Struct. Mol. Biol. 2011, 18, 177–185.
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone Deacetylases 1 and 2 Redundantly Regulate Cardiac Morphogenesis, Growth, and Contractility. Genes Dev. 2007, 21, 1790–1802.
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylases 1 and 2 Regulate Autophagy Flux and Skeletal Muscle Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654.
- Emmett, M.J.; Lazar, M.A. Integrative Regulation of Physiology by Histone Deacetylase 3. Nat. Rev. Mol. Cell Biol. 2018, 20, 102–115.
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587.e14.
- Lewandowski, S.L.; Janardhan, H.P.; Smee, K.M.; Bachman, M.; Sun, Z.; Lazar, M.A.; Trivedi, C.M. Histone Deacetylase 3 Modulates Tbx5 Activity to Regulate Early Cardiogenesis. Hum. Mol. Genet. 2014, 23, 3801–3809.
- Lewandowski, S.L.; Janardhan, H.P.; Trivedi, C.M. Histone Deacetylase 3 Coordinates Deacetylase-Independent Epigenetic Silencing of Transforming Growth Factor-Β1 (TGF-Β1) to Orchestrate Second Heart Field Development. J. Biol. Chem. 2015, 290, 27067–27089.
- Hong, S.; Zhou, W.; Fang, B.; Lu, W.; Loro, E.; Damle, M.; Ding, G.; Jager, J.; Zhang, S.; Zhang, Y.; et al. Dissociation of Muscle Insulin Sensitivity from Exercise Endurance in Mice by HDAC3 Depletion. Nat. Med. 2017, 23, 223–234.
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Gil-Rodríguez, M.C.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-Function HDAC8 Mutations Cause a Phenotypic Spectrum of Cornelia de Lange Syndrome-like Features, Ocular Hypertelorism, Large Fontanelle and X-Linked Inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900.
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-Dependent Nuclear Export of a Histone Deacetylase Regulates Muscle Differentiation. Nature 2000, 408, 106–111.
- Walkinshaw, D.R.; Weist, R.; Kim, G.-W.; You, L.; Xiao, L.; Nie, J.; Li, C.S.; Zhao, S.; Xu, M.; Yang, X.-J. The Tumor Suppressor Kinase LKB1 Activates the Downstream Kinases SIK2 and SIK3 to Stimulate Nuclear Export of Class IIa Histone Deacetylases. J. Biol. Chem. 2013, 288, 9345–9362.
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Identification of a Signal-Responsive Nuclear Export Sequence in Class II Histone Deacetylases. Mol. Cell. Biol. 2001, 21, 6312–6321.
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the Hidden Catalytic Activity of Vertebrate Class IIa Histone Deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340.
- Renzini, A.; Marroncelli, N.; Cavioli, G.; Di Francescantonio, S.; Forcina, L.; Lambridis, A.; Di Giorgio, E.; Valente, S.; Mai, A.; Brancolini, C.; et al. Cytoplasmic HDAC4 Regulates the Membrane Repair Mechanism in Duchenne Muscular Dystrophy. J. Cachexia. Sarcopenia Muscle 2022, 13, 1339–1359.
- Bruneteau, G.; Simonet, T.; Bauché, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.-L.; Behin, A.; Khiami, F.; Sariali, E.; et al. Muscle Histone Deacetylase 4 Upregulation in Amyotrophic Lateral Sclerosis: Potential Role in Reinnervation Ability and Disease Progression. Brain 2013, 136, 2359–2368.
- Pigna, E.; Simonazzi, E.; Sanna, K.; Bernadzki, K.M.; Proszynski, T.; Heil, C.; Palacios, D.; Adamo, S.; Moresi, V. Histone Deacetylase 4 Protects from Denervation and Skeletal Muscle Atrophy in a Murine Model of Amyotrophic Lateral Sclerosis. EBioMedicine 2019, 40, 717–732.
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 Deacetylase Associates with and Represses the MEF2 Transcription Factor. EMBO J. 1999, 18, 5099–5107.
- Davis, F.J.; Gupta, M.; Camoretti-Mercado, B.; Schwartz, R.J.; Gupta, M.P. Calcium/Calmodulin-Dependent Protein Kinase Activates Serum Response Factor Transcription Activity by Its Dissociation from Histone Deacetylase, HDAC4: IMPLICATIONS IN CARDIAC MUSCLE GENE REGULATION DURING HYPERTROPHY. J. Biol. Chem. 2003, 278, 20047–20058.
- Pigna, E.; Renzini, A.; Greco, E.; Simonazzi, E.; Fulle, S.; Mancinelli, R.; Moresi, V.; Adamo, S. HDAC4 Preserves Skeletal Muscle Structure Following Long-Term Denervation by Mediating Distinct Cellular Responses. Skelet. Muscle 2018, 8, 6.
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylase Degradation and MEF2 Activation Promote the Formation of Slow-Twitch Myofibers. J. Clin. Investig. 2007, 117, 2459–2467.
- Renzini, A.; Marroncelli, N.; Noviello, C.; Moresi, V.; Adamo, S. HDAC4 Regulates Skeletal Muscle Regeneration via Soluble Factors. Front. Physiol. 2018, 9, 1387.
- Marroncelli, N.; Bianchi, M.; Bertin, M.; Consalvi, S.; Saccone, V.; De Bardi, M.; Puri, P.L.; Palacios, D.; Adamo, S.; Moresi, V. HDAC4 Regulates Satellite Cell Proliferation and Differentiation by Targeting P21 and Sharp1 Genes. Sci. Rep. 2018, 8, 3448.
- Cohen, T.J.; Choi, M.-C.; Kapur, M.; Lira, V.A.; Yan, Z.; Yao, T.-P. HDAC4 Regulates Muscle Fiber Type-Specific Gene Expression Programs. Mol. Cells 2015, 38, 343.
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and Class II HDACs Control Neurogenic Muscle Atrophy by Inducing E3 Ubiquitin Ligases. Cell 2010, 143, 35–45.
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 Delays ALS Progression and Promotes Regeneration of Neuromuscular Synapses in Mice. Science 2009, 326, 1549–1554.
- Choi, M.-C.; Cohen, T.J.; Barrientos, T.; Wang, B.; Li, M.; Simmons, B.J.; Yang, J.S.; Cox, G.A.; Zhao, Y.; Yao, T.-P. A Direct HDAC4-MAP Kinase Crosstalk Activates Muscle Atrophy Program. Mol. Cell 2012, 47, 122–132.
- Luo, L.; Martin, S.C.; Parkington, J.; Cadena, S.M.; Zhu, J.; Ibebunjo, C.; Summermatter, S.; Londraville, N.; Patora-Komisarska, K.; Widler, L.; et al. HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1α, and Hsc70. Cell Rep. 2019, 29, 749–763.e12.
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; He, T.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. A Proteolytic Fragment of Histone Deacetylase 4 Protects the Heart from Failure by Regulating the Hexosamine Biosynthetic Pathway. Nat. Med. 2017, 24, 62–72.
- Jebessa, Z.H.; Shanmukha, K.D.; Dewenter, M.; Lehmann, L.H.; Xu, C.; Schreiter, F.; Siede, D.; Gong, X.-M.; Worst, B.C.; Federico, G.; et al. The Lipid-Droplet-Associated Protein ABHD5 Protects the Heart through Proteolysis of HDAC4. Nat. Metab. 2019, 1, 1157–1167.
- Klymenko, O.; Brecklinghaus, T.; Dille, M.; Springer, C.; de Wendt, C.; Altenhofen, D.; Binsch, C.; Knebel, B.; Scheller, J.; Hardt, C.; et al. Histone Deacetylase 5 Regulates Interleukin 6 Secretion and Insulin Action in Skeletal Muscle. Mol. Metab. 2020, 42, 101062.
- Harris, L.G.; Wang, S.H.; Mani, S.K.; Kasiganesan, H.; Chou, C.J.; Menick, D.R. Evidence for a Non-Canonical Role of HDAC5 in Regulation of the Cardiac Ncx1 and Bnp Genes. Nucleic Acids Res. 2016, 44, 3610–3617.
- Li Zhang, C.; McKinsey, T.A.; Olson, E.N. The Transcriptional Corepressor MITR Is a Signal-Responsive Inhibitor of Myogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7354–7359.
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II Histone Deacetylases Act as Signal-Responsive Repressors of Cardiac Hypertrophy. Cell 2002, 110, 479–488.
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178.
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.W.; Khochbin, S. HDAC6-P97/VCP Controlled Polyubiquitin Chain Turnover. EMBO J. 2006, 25, 3357–3366.
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458.
- Osseni, A.; Ravel-Chapuis, A.; Thomas, J.-L.; Gache, V.; Schaeffer, L.; Jasmin, B.J. HDAC6 Regulates Microtubule Stability and Clustering of AChRs at Neuromuscular Junctions. J. Cell Biol. 2020, 219, e201901099.
- Ratti, F.; Ramond, F.; Moncollin, V.; Simonet, T.; Milan, G.; Méjat, A.; Thomas, J.L.; Streichenberger, N.; Gilquin, B.; Matthias, P.; et al. Histone Deacetylase 6 Is a FoxO Transcription Factor-Dependent Effector in Skeletal Muscle Atrophy. J. Biol. Chem. 2015, 290, 4215–4224.
- Lin, Y.H.; Major, J.L.; Liebner, T.; Hourani, Z.; Travers, J.G.; Wennersten, S.A.; Haefner, K.R.; Cavasin, M.A.; Wilson, C.E.; Jeong, M.Y.; et al. HDAC6 Modulates Myofibril Stiffness and Diastolic Function of the Heart. J. Clin. Investig. 2022, 132, e148333.
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone Deacetylase 10 Structure and Molecular Function as a Polyamine Deacetylase. Nat. Commun. 2017, 8, 15368.
- Michan, S.; Sinclair, D. Sirtuins in Mammals: Insights into Their Biological Function. Biochem. J. 2007, 404, 1–13.
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794.
- Planavila, A.; Dominguez, E.; Navarro, M.; Vinciguerra, M.; Iglesias, R.; Giralt, M.; Lope-Piedrafita, S.; Ruberte, J.; Villarroya, F. Dilated Cardiomyopathy and Mitochondrial Dysfunction in Sirt1-Deficient Mice: A Role for Sirt1-Mef2 in Adult Heart. J. Mol. Cell. Cardiol. 2012, 53, 521–531.
- Wang, Y.; Xu, C.; Liang, Y.; Vanhoutte, P.M. SIRT1 in Metabolic Syndrome: Where to Target Matters. Pharmacol. Ther. 2012, 136, 305–318.
- Bai, B.; Vanhoutte, P.M.; Wang, Y. Loss-of-SIRT1 Function during Vascular Ageing: Hyperphosphorylation Mediated by Cyclin-Dependent Kinase 5. Trends Cardiovasc. Med. 2014, 24, 81–84.
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats. PLoS ONE 2011, 6, e191942011.
- Lee, D.; Goldberg, A.L. SIRT1 Protein, by Blocking the Activities of Transcription Factors FoxO1 and FoxO3, Inhibits Muscle Atrophy and Promotes Muscle Growth. J. Biol. Chem. 2013, 288, 30515–30526.
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK Regulates Energy Expenditure by Modulating NAD+ Metabolism and SIRT1 Activity. Nature 2009, 458, 1056–1060.
- Tian, H.; Liu, S.; Ren, J.; Lee, J.K.W.; Wang, R.; Chen, P. Role of Histone Deacetylases in Skeletal Muscle Physiology and Systemic Energy Homeostasis: Implications for Metabolic Diseases and Therapy. Front. Physiol. 2020, 11, 949.
- Simmons, B.J.; Cohen, T.J.; Bedlack, R.; Yao, T.P. HDACs in Skeletal Muscle Remodeling and Neuromuscular Disease. Handb. Exp. Pharmacol. 2011, 206, 79–101.
- Iezzi, S.; Di Padova, M.; Serra, C.; Caretti, G.; Simone, C.; Maklan, E.; Minetti, G.; Zhao, P.; Hoffman, E.P.; Puri, P.L.; et al. Deacetylase Inhibitors Increase Muscle Cell Size by Promoting Myoblast Recruitment and Fusion through Induction of Follistatin. Dev. Cell 2004, 6, 673–684.
- Rajendran, P.; Williams, D.E.; Ho, E.; Dashwood, R.H. Metabolism as a Key to Histone Deacetylase Inhibition. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 181.
- Song, S.; Wen, Y.; Tong, H.; Loro, E.; Gong, Y.; Liu, J.; Hong, S.; Li, L.; Khurana, T.S.; Chu, M.; et al. The HDAC3 Enzymatic Activity Regulates Skeletal Muscle Fuel Metabolism. J. Mol. Cell Biol. 2019, 11, 133–143.
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Control of Muscle Development by Dueling HATs and HDACs. Curr. Opin. Genet. Dev. 2001, 11, 497–504.
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) Regulates Skeletal Muscle Metabolism and Insulin Signaling via Altered Mitochondrial Oxidation and Reactive Oxygen Species Production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613.
- Vargas-Ortiz, K.; Pérez-Vázquez, V.; Macías-Cervantes, M.H. Exercise and Sirtuins: A Way to Mitochondrial Health in Skeletal Muscle. Int. J. Mol. Sci. 2019, 20, 2717.
- McGee, S.L.; Hargreaves, M. Histone Modifications and Exercise Adaptations. J. Appl. Physiol. 2011, 110, 258–263.
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038.
- Lovering, R.M.; Porter, N.C.; Block, R.J. The Muscular Dystrophies: From Genes to Therapies. Phys. Ther. 2005, 85, 1372.
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13.
- Ervasti, J.M. Dystrophin, Its Interactions with Other Proteins, and Implications for Muscular Dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117.
- Ehmsen, J.; Poon, E.; Davies, K. The Dystrophin-Associated Protein Complex. J. Cell Sci. 2002, 115, 2801–2803.
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric Oxide Synthase Complexed with Dystrophin and Absent from Skeletal Muscle Sarcolemma in Duchenne Muscular Dystrophy. Cell 1995, 82, 743–752.
- Vandebrouck, A.; Sabourin, J.; Rivet, J.; Balghi, H.; Sebille, S.; Kitzis, A.; Raymond, G.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of Capacitative Calcium Entries by A1-syntrophin: Association of TRPC1 with Dystrophin Complex and the PDZ Domain of A1-syntrophin. FASEB J. 2007, 21, 608–617.
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2015, 96, 253–305.
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal Models of Duchenne Muscular Dystrophy: From Basic Mechanisms to Gene Therapy. Dis. Model. Mech. 2015, 8, 195–213.
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short Telomeres and Stem Cell Exhaustion Model Duchenne Muscular Dystrophy in Mdx/MTR Mice. Cell 2010, 143, 1059–1071.
- McDonald, A.A.; Hebert, S.L.; Kunz, M.D.; Ralles, S.J.; McLoon, L.K. Disease Course in Mdx:Utrophin+/− Mice: Comparison of Three Mouse Models of Duchenne Muscular Dystrophy. Physiol. Rep. 2015, 3, e123912015.
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tomé, F.M.S.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and Developmental Expression of Dystrophin Related Protein in Skeletal Muscle. Neuromuscul. Disord. 1991, 1, 185–194.
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-Dystrophin-Deficient Mice as a Model for Duchenne Muscular Dystrophy. Cell 1997, 90, 717–727.
- Soblechero-Martín, P.; López-Martínez, A.; de la Puente-Ovejero, L.; Vallejo-Illarramendi, A.; Arechavala-Gomeza, V. Utrophin Modulator Drugs as Potential Therapies for Duchenne and Becker Muscular Dystrophies. Neuropathol. Appl. Neurobiol. 2021, 47, 711.
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.M.; Davies, K. Expression of Full-Length Utrophin Prevents Muscular Dystrophy in Mdx Mice. Nat. Med. 1998, 4, 1441–1444.
- Barton, E.R. Impact of Sarcoglycan Complex on Mechanical Signal Transduction in Murine Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2006, 290.
- Tarakci, H.; Berger, J. The Sarcoglycan Complex in Skeletal Muscle. Front. Biosci. 2016, 21, 744–756.
- Angelini, C. LGMD. Identification, Description and Classification. Acta Myol. 2020, 39, 207–217.
- Vainzof, M.; Passos-Bueno, M.R.; Canovas, M.; Moreira, E.S.; Pavanello, R.C.M.; Marie, S.K.; Anderson, L.V.B.; Bonnemann, C.G.; McNally, E.M.; Nigro, V.; et al. The Sarcoglycan Complex in the Six Autosomal Recessive Limb-Girdle Muscular Dystrophies. Hum. Mol. Genet. 1996, 5, 1963–1969.
- Kirschner, J.; Lochmüller, H. Sarcoglycanopathies. Handb. Clin. Neurol. 2011, 101, 41–46.
- Sandonà, D.; Betto, R. Sarcoglycanopathies: Molecular Pathogenesis and Therapeutic Prospects. Expert Rev. Mol. Med. 2009, 11, e28.
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 Blockade by Nitric Oxide and Histone Deacetylase Inhibitors Reveals a Common Target in Duchenne Muscular Dystrophy Treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187.
- Colussi, C.; Gurtner, A.; Rosati, J.; Illi, B.; Ragone, G.; Piaggio, G.; Moggio, M.; Lamperti, C.; D’Angelo, G.; Clementi, E.; et al. Nitric Oxide Deficiency Determines Global Chromatin Changes in Duchenne Muscular Dystrophy. FASEB J. 2009, 23, 2131–2141.
- Tidball, J.G.; Wehling-Henricks, M. Nitric Oxide Synthase Deficiency and the Pathophysiology of Muscular Dystrophy. J. Physiol. 2014, 592, 4627–4638.
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs Involved in Molecular Circuitries Relevant for the Duchenne Muscular Dystrophy Pathogenesis Are Controlled by the Dystrophin/NNOS Pathway. Cell Metab. 2010, 12, 341–351.
- Abe, S.; Soejima, M.; Iwanuma, O.; Saka, H.; Matsunaga, S.; Sakiyama, K.; Ide, Y. Expression of Myostatin and Follistatin in Mdx Mice, an Animal Model for Muscular Dystrophy. Zoolog. Sci. 2009, 26, 315–320.
- Bogdanovich, S.; Krag, T.O.B.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional Improvement of Dystrophic Muscle by Myostatin Blockade. Nature 2002, 420, 418–421.
- Parsons, S.A.; Millay, D.P.; Sargent, M.A.; McNally, E.M.; Molkentin, J.D. Age-Dependent Effect of Myostatin Blockade on Disease Severity in a Murine Model of Limb-Girdle Muscular Dystrophy. Am. J. Pathol. 2006, 168, 1975–1985.
- Madej-Pilarczyk, A. Clinical Aspects of Emery-Dreifuss Muscular Dystrophy. Nucleus 2018, 9, 314–320.
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a Novel X-Linked Gene Responsible for Emery-Dreifuss Muscular Dystrophy. Nat. Genet. 1994, 8, 323–327.
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The Nuclear Envelope Protein Emerin Binds Directly to Histone Deacetylase 3 (HDAC3) and Activates HDAC3 Activity. J. Biol. Chem. 2012, 287, 22080–22088.
- Demmerle, J.; Koch, A.J.; Holaska, J.M. Emerin and Histone Deacetylase 3 (HDAC3) Cooperatively Regulate Expression and Nuclear Positions of MyoD, Myf5, and Pax7 Genes during Myogenesis. Chromosome Res. 2013, 21, 765–779.
- Collins, C.M.; Ellis, J.A.; Holaska, J.M. MAPK Signaling Pathways and HDAC3 Activity Are Disrupted during Differentiation of Emerin-Null Myogenic Progenitor Cells. Dis. Model. Mech. 2017, 10, 385.
- Spreafico, M.; Cafora, M.; Bragato, C.; Capitanio, D.; Marasca, F.; Bodega, B.; De Palma, C.; Mora, M.; Gelfi, C.; Marozzi, A.; et al. Targeting HDAC8 to Ameliorate Skeletal Muscle Differentiation in Duchenne Muscular Dystrophy. Pharmacol. Res. 2021, 170, 105750.
- Osseni, A.; Ravel-Chapuis, A.; Scionti, I.; Gangloff, Y.-G.; Moncollin, V.; Mounier, R.; Leblanc, P.; Jasmin, B.J.; Schaeffer, L. Pharmacological Inhibition of HDAC6 Downregulates TGF-β via Smad2/3 Acetylation and Improves Dystrophin-Deficient Muscles. bioRxiv 2022.
- Agrawal, A.; Clayton, E.L.; Cavazos, C.L.; Clayton, B.A.; Rodney, G.G. Histone Deacetylase 6 Inhibition Promotes Microtubule Acetylation and Facilitates Autophagosome-Lysosome Fusion in Dystrophin-Deficient Mdx Mice. bioRxiv 2022.
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738.
- Wagner, S.; Manickam, R.; Brotto, M.; Tipparaju, S.M. NAD+ Centric Mechanisms and Molecular Determinants of Skeletal Muscle Disease and Aging. Mol. Cell. Biochem. 2022, 477, 1829–1848.
- Hardee, J.P.; Caldow, M.K.; Chan, A.S.M.; Plenderleith, S.K.; Trieu, J.; Koopman, R.; Lynch, G.S. Dystrophin Deficiency Disrupts Muscle Clock Expression and Mitochondrial Quality Control in Mdx Mice. Am. J. Physiol. Cell Physiol. 2021, 321, C288–C296.
- Hulmi, J.J.; Hentilä, J.; DeRuisseau, K.C.; Oliveira, B.M.; Papaioannou, K.G.; Autio, R.; Kujala, U.M.; Ritvos, O.; Kainulainen, H.; Korkmaz, A.; et al. Effects of Muscular Dystrophy, Exercise and Blocking Activin Receptor IIB Ligands on the Unfolded Protein Response and Oxidative Stress. Free Radic. Biol. Med. 2016, 99, 308–322.
- Chalkiadaki, A.; Igarashi, M.; Nasamu, A.S.; Knezevic, J.; Guarente, L. Muscle-Specific SIRT1 Gain-of-Function Increases Slow-Twitch Fibers and Ameliorates Pathophysiology in a Mouse Model of Duchenne Muscular Dystrophy. PLoS Genet. 2014, 10, e1004490.
- Domi, E.; Hoxha, M.; Prendi, E.; Zappacosta, B. A Systematic Review on the Role of SIRT1 in Duchenne Muscular Dystrophy. Cells 2021, 10, 1380.
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1alpha Regulates the Neuromuscular Junction Program and Ameliorates Duchenne Muscular Dystrophy. Genes Dev. 2007, 21, 770–783.
- Spaulding, H.R.; Ludwig, A.K.; Hollinger, K.; Hudson, M.B.; Selsby, J.T. PGC-1α Overexpression Increases Transcription Factor EB Nuclear Localization and Lysosome Abundance in Dystrophin-Deficient Skeletal Muscle. Physiol. Rep. 2020, 8, e14383.
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of Dystrophic Skeletal Muscle by PGC-1α Involves a Fast to Slow Fiber Type Shift in the Mdx Mouse. PLoS ONE 2012, 7, e0030063.
- Fujiwara, D.; Iwahara, N.; Sebori, R.; Hosoda, R.; Shimohama, S.; Kuno, A.; Horio, Y. SIRT1 Deficiency Interferes with Membrane Resealing after Cell Membrane Injury. PLoS ONE 2018, 14, e0218329.
- Han, Z.; Chang, C.; Zhu, W.; Zhang, Y.; Zheng, J.; Kang, X.; Jin, G.; Gong, Z. Role of SIRT2 in Regulating the Dexamethasone-Activated Autophagy Pathway in Skeletal Muscle Atrophy. Biochem. Cell Biol. 2021, 99, 562–569.
- Wu, G.; Song, C.; Lu, H.; Jia, L.; Yang, G.; Shi, X.; Sun, S. Sirt2 Induces C2C12 Myoblasts Proliferation by Activation of the ERK1/2 Pathway. Acta Biochim. Biophys. Sin. 2014, 46, 342–345.
- Lee, E.J.; Lee, M.M.; Park, S.Y.; Jeong, K.S. Sirt2 Positively Regulates Muscle Regeneration after Notexin-Induced Muscle Injury. Exp. Mol. Pathol. 2022, 127, 104798.
- Georgieva, A.M.; Guo, X.; Bartkuhn, M.; Günther, S.; Künne, C.; Smolka, C.; Atzberger, A.; Gärtner, U.; Mamchaoui, K.; Bober, E.; et al. Inactivation of Sirt6 Ameliorates Muscular Dystrophy in Mdx Mice by Releasing Suppression of Utrophin Expression. Nat. Commun. 2022, 13, 4184.
- Guiraud, S.; Edwards, B.; Squire, S.E.; Babbs, A.; Shah, N.; Berg, A.; Chen, H.; Davies, K.E. Identification of Serum Protein Biomarkers for Utrophin Based DMD Therapy. Sci. Rep. 2017, 7, 43697.
- Budzinska, M.; Zimna, A.; Kurpisz, M. The Role of Mitochondria in Duchenne Muscular Dystrophy. J. Physiol. Pharmacol. 2021, 72, 157–166.
- Mocciaro, E.; Runfola, V.; Ghezzi, P.; Pannese, M.; Gabellini, D. DUX4 Role in Normal Physiology and in FSHD Muscular Dystrophy. Cells 2021, 10, 3322.
- Cui, X.; Yao, L.; Yang, X.; Gao, Y.; Fang, F.; Zhang, J.; Wang, Q.; Chang, Y. SIRT6 Regulates Metabolic Homeostasis in Skeletal Muscle through Activation of AMPK. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E493–E505.
- Samant, S.A.; Kanwal, A.; Pillai, V.B.; Bao, R.; Gupta, M.P. The Histone Deacetylase SIRT6 Blocks Myostatin Expression and Development of Muscle Atrophy. Sci. Rep. 2017, 7, 11877.
- Moreno-Yruela, C.; Galleano, I.; Madsen, A.S.; Olsen, C.A. Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol. 2018, 25, 849–856.e8.
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693.
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 Regulates Type I Interferon Signaling through Defatty-Acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492.
- Núñez-Álvarez, Y.; Hurtado, E.; Muñoz, M.; García-Tuñon, I.; Rech, G.E.; Pluvinet, R.; Sumoy, L.; Pendás, A.M.; Peinado, M.A.; Suelves, M. Loss of HDAC11 Accelerates Skeletal Muscle Regeneration in Mice. FEBS J. 2021, 288, 1201–1223.
- Núñez-Álvarez, Y.; Suelves, M. HDAC11: A Multifaceted Histone Deacetylase with Proficient Fatty Deacylase Activity and Its Roles in Physiological Processes. FEBS J. 2022, 289, 2771–2792.
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 Reduces the Pathology of Mdx Muscular Dystrophy by Deactivating M1 Macrophages and Modulating Macrophage Phenotype. Hum. Mol. Genet. 2011, 20, 790–805.
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Chiyo, T.; Nishiyama, A.; Okada, H.; Takeda, S.; Okada, T. Dystrophic Mdx Mice Develop Severe Cardiac and Respiratory Dysfunction Following Genetic Ablation of the Anti-Inflammatory Cytokine IL-10. Hum. Mol. Genet. 2014, 23, 3990–4000.
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. Mol. Med. 2011, 17, 457.
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; Del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical Studies in the Mdx Mouse Model of Duchenne Muscular Dystrophy with the Histone Deacetylase Inhibitor Givinostat. Mol. Med. 2013, 19, 79–87.
- Sandoná, M.; Consalvi, S.; Tucciarone, L.; Puri, P.L.; Saccone, V. HDAC Inhibitors for Muscular Dystrophies: Progress and Prospects. Expert Opin. Orphan Drugs 2016, 4, 125–127.
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326.
- Iezzi, S.; Cossu, G.; Nervi, C.; Sartorelli, V.; Puri, P.L. Stage-Specific Modulation of Skeletal Myogenesis by Inhibitors of Nuclear Deacetylases. Proc. Natl. Acad. Sci. USA 2002, 99, 7757–7762.
- Consalvi, S.; Tucciarone, L.; Macrì, E.; De Bardi, M.; Picozza, M.; Salvatori, I.; Renzini, A.; Valente, S.; Mai, A.; Moresi, V.; et al. Determinants of Epigenetic Resistance to HDAC Inhibitors in Dystrophic Fibro-Adipogenic Progenitors. EMBO Rep. 2022, 23, e54721.
- Mozzetta, C.; Consalvi, S.; Saccone, V.; Tierney, M.; Diamantini, A.; Mitchell, K.J.; Marazzi, G.; Borsellino, G.; Battistini, L.; Sassoon, D.; et al. Fibroadipogenic Progenitors Mediate the Ability of HDAC Inhibitors to Promote Regeneration in Dystrophic Muscles of Young, but Not Old Mdx Mice. EMBO Mol. Med. 2013, 5, 626–639.
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandoná, M.; Ryan, T.; Rojas-Muñoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-Regulated MyomiRs Control BAF60 Variant Exchange and Direct the Functional Phenotype of Fibro-Adipogenic Progenitors in Dystrophic Muscles. Genes Dev. 2014, 28, 841–857.
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle Injury Activates Resident Fibro/Adipogenic Progenitors That Facilitate Myogenesis. Nat. Cell Biol. 2010, 12, 153–163.
- Uezumi, A.; Fukada, S.I.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal Progenitors Distinct from Satellite Cells Contribute to Ectopic Fat Cell Formation in Skeletal Muscle. Nat. Cell Biol. 2010, 12, 143–152.
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and Adipogenesis Originate from a Common Mesenchymal Progenitor in Skeletal Muscle. J. Cell Sci. 2011, 124, 3654–3664.
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 Positively Regulates Autophagy and Mitochondria Function in Embryonic Stem Cells under Oxidative Stress. Stem Cells 2014, 32, 1183–1194.
- In, H.L.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A Role for the NAD-Dependent Deacetylase Sirt1 in the Regulation of Autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379.
- Ghosh, A.K.; Varga, J. The Transcriptional Coactivator and Acetyltransferase P300 in Fibroblast Biology and Fibrosis. J. Cell. Physiol. 2007, 213, 663–671.
More