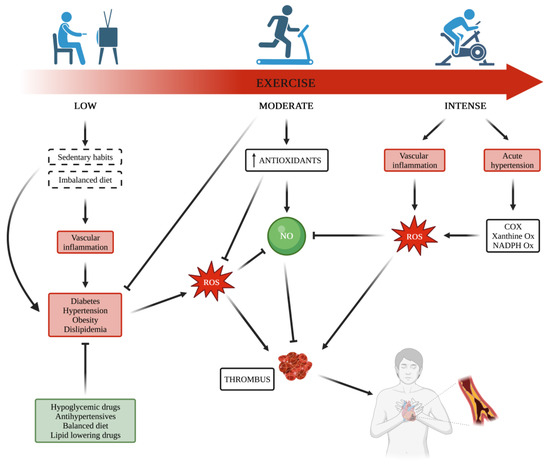
2. Metabolic Diseases and Exercise Effects on Platelets
Metabolic disorders, including obesity, dyslipidemia, and diabetes are well established risk factors for CVDs
[16][145], being associated with underlying abnormalities that trigger some common biochemical events influencing platelet response, such as enhanced ROS formation, decreased availability and/or synthesis of NO, lipid peroxidation with the consequent increased production of TXA
2 and free radicals, and non-enzymatic–catalyzed generation of bioactive isoprostanes, which activate TX receptor
[11][17][18][11,146,147].
In each of these metabolic disorders, it is reasonable to suppose that physical activity, with or without weight reduction, reduces cardiometabolic disorder risk, partially by improving insulin sensitivity and lowering blood pressure
[19][20][148,149]. Indeed, among the biological mechanisms by which exercise confers its benefits, the increased sensitivity to both the metabolic
[21][22][150,151] and vascular
[23][152] actions of insulin may play an important role. Platelet membrane expresses insulin receptors with a density similar to that of cell types of the targets of the metabolic actions of the hormone
[24][153]. Platelet insulin receptors activate the intracellular pathway classically linked to insulin signaling, even in the absence of a response of glucose uptake
[25][154]. Obese insulin-resistant subjects with or without diabetes show impaired platelet responsiveness to the inhibitory effects of insulin
[26][27][25,155], thus suggesting that each strategy useful in reducing insulin-resistance can also improve the vascular effects of insulin including its antiplatelet effects.
Platelet Alterations in Obesity. One of the first epidemiological studies showing a strong correlation between obesity and CV events was the Framingham Heart Study
[28][29][156,157]. From then, many other studies have confirmed the role of waist-to-hip ratio (WHR), an index of central obesity, as the strongest anthropometric predictor of myocardial infarction
[30][158] and stroke
[31][32][159,160].
The release of cytokines and free fatty acids from abdominal adiposity has a causal, unfavorable effect on lipid profile and other cardiometabolic risk factors involved in the pathogenesis of both atherothrombosis and insulin resistance
[33][34][161,162]. Chronic low-grade inflammation and systemic oxidative stress have both been associated with obesity causing endothelial dysfunction with the consequent loss of its antithrombotic properties and arterial damage, thus justifying the assumption that obesity is a pro-thrombotic condition due to vascular disease, increased platelet activation, and hypercoagulability
[35][36][37][38][39][163,164,165,166,167]. Adipose tissue is an important source of ROS and the increased level of systemic oxidative stress contributes to the development of obesity-associated insulin resistance and type 2 diabetes mellitus (T2DM) and other disorders, such as hypertension, atherosclerosis, and cancer
[40][41][168,169]. Excess intake of nutrients, a sedentary lifestyle, and the consequent weight gain promote ROS production and mitochondrial dysfunction
[42][170], a risk factor for T2DM, atherosclerosis, and hypertension
[43][171]. Among the in vivo parameters of platelet activation, MPV, a marker closely related to platelet hyperactivation, has been found to be increased in obesity
[44][45][46][17,172,173], and a positive correlation also exists between MPV and body mass index (BMI) after weight loss
[45][47][48][172,174,175]. A platelet activation marker associated with obesity is also sP-sel, which is able to predict atherosclerosis independently of BMI and other CVD risk factors
[49][176]. The increased circulating levels of sP-sel found in overweight and obese subjects
[11][50][11,177] are reduced after weight loss
[11]. Obese subjects show increased levels of 11-dehydro-TXB
2 and PGF
2α, thus underlining the link between platelet activation and oxidative stress
[51][178]. Indeed, the chronic ‘metabolic inflammation,’ which is considered the hallmark of obesity and causes insulin resistance and T2DM
[52][179], significantly contributes to increases in the systemic levels of ROS, which affect platelet reactivity by different mechanisms, including decreased NO bioavailability, increased expression of membrane glycoproteins, impairment of calcium mobilization, and isoprostane generation
[51][178]. In obesity, in comparison with non-obese subjects, elevated PMP levels positively correlate with BMI and waist circumference
[53][180], although this finding was not confirmed in other studies, where PMP did not appear to differ in number
[54][181] but were greatly heterogeneous in size and distribution, with different levels of proteins involved in thrombosis and tumorigenesis
[54][181].
In previous studies, the researchers provided evidence of persistent platelet hyporesponsiveness to NO and PGI
2 pathways in obesity and T2DM
[55][56][57][26,182,183]. Researchers demonstrated the presence of multi-step defects at the level of NO/cGMP/PKG and PGI
2/cAMP/PKA pathways. Specifically, platelets from obese subjects show an impairment in the respective abilities of NO and PGI
2 to increase cGMP and cAMP synthesis, and resistance of cGMP and cAMP themselves in activating their specific kinases PKG and PKA
[55][56][26,182]. As these are cyclic nucleotides effective in reducing intracellular Ca
2+ [58][184], the data explained one of the mechanisms implicated in Ca
2+ flux alterations found in insulin-resistance states
[59][185] and the defective action of cyclic nucleotides on platelet function. In addition, hyperglycemia does not emphasize this multistep resistance
[60][186], and the presence of diabetes without obesity is not associated with platelet abnormalities observed in obese subjects
[60][186]. These findings support the hypothesis that the abnormalities leading to platelet hyperreactivity are mainly related to the underlying metabolic disorders dependent on visceral adipose tissue activity rather than on platelet exposure to hyperglycemia effects. Besides changing subcutaneous and visceral adipose tissue distribution, insulin sensitivity, and beta-cell performance, a dietary program aiming at achieving weight loss of at least 7–10% of initial body weight leads to a significant reduction in systemic inflammation, oxidative stress, lipid peroxidation, and platelet reactivity
[11].
Exercise Effects on Platelets in Obesity. A recent systematic review and meta-analysis including 25 randomized controlled trials (1686 participants) shows that regular aerobic exercise significantly decreases visceral adipose tissue with more pronounced benefits for higher intensity exercise
[61][187]. It has also been ascertained that independently of age, body mass index, and exercise training characteristics, aerobic training in adults with overweightness or obesity and with cardiometabolic disorders is effective in reducing postprandial glucose and insulin levels
[62][188]. As far as platelet parameters are concerned, a randomized clinical trial performed in overweight men showed that moderate-intensity training for 12 weeks consisting in walking/slow jogging exercise at 45–55% of VO
2 max (5x/week for 45–60 min) led to a reduction in platelet aggregation associated with a reduction in serum TXB
2 levels
[63][135]. To determine the role of exercise in platelet reactivity in obese patients with coronary artery disease, a 4-month program of training exercise and behavioral weight loss was performed by Keating et al.
[64][189]. These authors found a significant decrease in P-sel expression not independently associated with measures of body composition or fitness. After controlling for exercise group and gender, the change in platelet reactivity was more pronounced in females and associated with changes in high-sensitivity C-reactive protein and a reduction in insulin-resistance. A study carried out on blood samples taken from obese women before and immediately after exercise demonstrated that vigorous aerobic exercise, consisting of a 30-min walking exercise test at an intensity of 70% of individual peak oxygen uptake, was able to significantly prolong the clot formation time as measured by thromboelastometry and reduce the fibrin buildup after exercise. Thrombography revealed a significant exercise-induced decrease in endogenous thrombin potential
[65][121]. On the basis of these results, the authors postulated that vigorous aerobic exercise might be a suitable strategy to protect obese women from thrombotic events. Indeed, this assertion has been confuted in favor of regular exposure to high-intensity exercise in order to desensitize against exercise-induced platelet aggregation, attenuate coagulatory parameters, and up-regulate fibrinolytic potential
[66][87]. In another study, obese subjects underwent moderate-intensity exercise on a treadmill (at 60% of their VO
2 max), and the results showed changes in size distribution and cell origin of extracellular vesicles (EVs)
[67][190]. Total EVs, exosomes, and CD61+ EVs were significantly associated with HOMA-IR, and flow cytometry assays revealed that acute exercise provided a significant improvement of hemostasis parameters, including reduced platelet aggregability
[68][191] (
Table 1).
Platelet Alterations in Dyslipidemia. Dyslipidemia promotes the atherosclerosis process because of the chronic accumulation of lipid-rich plaque in arteries
[69][192], and its relationship with the increase in CV risk depends on its long-term effects on atherogenesis as well as on its influence on thrombogenesis
[70][28]. Lipid profile alterations are associated with increased oxidative stress, and the generation of oxidized lipids, such as ox-LDL, leading to platelet hyperreactivity
[71][72][16,193]. In turn, activated platelets can generate ox-LDL, thus contributing to propagating platelet activation, and inducing thrombus formation through oxidative stress-mediated mechanisms. In particular, the ROS-producing enzyme nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase (NOX)-2 (NOX2)-dependent increase in oxidative stress is involved in platelet ability to propagate the oxidation of lipoproteins
[73][74][194,195]. Oxidized lipoproteins are deeply involved in many biochemical events leading to atherosclerosis processes as well as platelet hyperactivation
[75][76][196,197].
Platelet hyperreactivity, strongly and independently associated with thrombotic events
[77][78][79][80][81][198,199,200,201,202], is characterized by redox imbalance
[9][10][82][9,10,203]. Mechanistically, ROS and oxidation reactions are, per se, the cause of vascular dysfunction
[40][83][84][168,204,205], but dyslipidemia also increases the risk of CVDs due to the effects of plasma oxidized lipids on platelet function
[85][206] via the interaction of ox-LDL with scavenger receptors, such as CD36, and signaling pathways, including the Src family kinases (SFK), mitogen-activated protein kinases (MAPK), and NADPH-oxidase.
In their native form, LDL particles do not increase platelet aggregation, whereas their oxidative modifications make these lipoproteins able to act as aggregating agents in the absence of physiological agonists
[86][207]. In hypercholesterolemia, the loss and/or impaired effects of NO on platelets are important determinants for platelet hyperactivation. The increased oxidative stress causes a decrease in NO bioavailability
[87][208] and a decreased sensitivity to NO-related pathways
[11][88][89][90][11,209,210,211]. Additionally, a lower NO-mediated inhibitory effect of the incretin hormone glucagon-like peptide 1(GLP-1)
[91][212] has been found as a putative mechanism by which hypercholesterolemia can induce platelet hyperactivation
[88][209]. Patients with hypercholesterolemia show increased levels of TXA
2, superoxide anion, and platelet activation markers, including sP-Sel, PF-4, sCD-40L, and β-TG
[9][10][82][9,10,203]. Importantly, the reductions in oxidative stress-related abnormalities obtained through pharmacological interventions can significantly improve platelet function. Indeed, many platelet alterations significantly improve after treatment with classical lipid-lowering drugs, such as statins
[10][92][93][94][95][96][10,81,213,214,215,216], and the more recent and aggressive therapies, such as the anti-proprotein convertase subtilisin/kexin type 9 (PCSK9) antibodies
[9][97][9,217].
Exercise, Lipid Metabolism, and Platelets. Exercise has positive impacts on reducing cholesterol levels and improving the physical fitness of individuals with dyslipidaemia
[98][218]. A significant positive association exists between exercise and HDL cholesterol, while a significant negative association has been reported between exercise and triglyceride levels, total cholesterol, LDL cholesterol, and triglycerides after a 5-year follow-up
[99][219]. It is generally accepted that regular physical exercise, with a linear dose–response relationship, increases HDL cholesterol while regulating and theoretically preventing increases in LDL cholesterol and triglycerides
[100][220]. More intense activity seems required to obtain significant reductions in LDL cholesterol and triglyceride levels, even if a bout of prolonged aerobic exercise has been shown to be effective in lowering blood postprandial hypertriglyceride levels in individuals at high risk of developing CVD
[101][221]. Indeed, exercise modality, cardiovascular exercise type, and timing of exercise can vary in their attenuation of postprandial triglyceride levels depending on exercise energy expenditure prior to meal administration
[102][222].
As known, exercise increases oxygen consumption with the consequent increase in oxygen-related free radicals. The increased exercise-mediated oxidative stress can induce lipid peroxidation, membrane damage, and platelet activation due to the effects of both native LDL and LDL modified under oxidative stress on platelets
[103][104][223,224]. The deleterious effect of strenuous exercise on platelet activity has been confirmed by a study carried out on healthy subjects showing an increase in plasma TX level at peak exercise and its return to pre-exercise levels at 10 min postexercise
[105][84]. Intriguingly, this study also showed that a treadmill exercise test to the point of physical exhaustion induced platelet aggregation, increased TX,
β-TG, and lipid peroxide levels. However, that acute exercise decreased LDL lipid peroxides, reaching a statistically significant lower plasma concentration at 10 min post-exercise. The authors speculated that during strenuous exercise, LDL lipid peroxides can replace plasma LDL cholesterol (LDL-C), attenuating the role of LDL on platelet activation. To explain this paradoxical result, ex vivo experiments were also performed by adding mildly oxidized LDL to peak exercise blood. The result was a decrease in platelet aggregation, suggesting that LDL lipid peroxides attenuate exercise-induced platelet aggregation. However, the question of why mildly ox-LDL, in conditions of strenuous exercise, attenuated instead of stimulating platelet aggregation remains to be explored.
In another study, sedentary individuals performing exercise training for 8 weeks failed to decrease their circulating ox-LDL levels but reduced plasma total cholesterol and LDL-C levels and positively influenced platelet function, as demonstrated by the reduced ability of ox-LDL added in vitro to increase the agonist-induced aggregation and intraplatelet calcium elevation in blood samples collected at both resting and postexercise
[106][75]. However, detraining reverses the benefits of training on lipid profile and platelet function, and in contrast to regular, strenuous acute exercise, it increases platelet aggregation and calcium elevation promoted by 100 microg/mL of ox-LDL
[106][75]. These findings confirm the positive effects of the adaptation to long-term exercise training.
Besides lipid profile amelioration, the high-fat diet combined with the swimming group is able to improve many hemostasis parameters, including platelet reactivity, as shown by prolonged bleeding time, reduced platelet aggregability and spread of fibrinogen, and decreased activation of pathways implicated in platelet activation
[107][225].
The effects of an 8-week high-intensity aerobic exercise on in vivo lipid peroxidation and platelet activation were investigated in healthy sedentary individuals with low HDL cholesterol levels. Exercise training did not modify total cholesterol or LDL-C concentrations but significantly reduced oxidative stress (8-iso-PGF
2α) and platelet activation (11-dehydro-TXB
2) urinary markers
[108][226] (
Table 1).
Platelet Alterations in Diabetes. Several studies provide evidence of the enhanced activation of platelets in T2DM
[109][110][111][112][113][227,228,229,230,231]. Increased values of MPV, an indicator of platelets larger in size and metabolically more active, and platelet distribution width (PDW) are indicative of platelet activation and associated with thrombotic events
[114][232]. The consequent platelet hyperreactivity triggers the release of multiple molecules stored in α-granules, dense granules, and lysosomal granules.
The persistent platelet activation represents an important link between diabetes and atherothrombosis, although evidence from the literature shows platelet activation already in prediabetes
[115][233] or newly diagnosed T2DM patients with central obesity in good metabolic control
[116][234]. Indeed, lipid peroxidation and TX-dependent platelet activation, as mirrored by in vivo urinary excretion of PGF
2α and 11-dehydro-TXB₂, correlate with atherothrombosis from the earlier stages of T2DM
[116][234]. Nevertheless, a linear correlation was observed between the urinary excretion of the stable TX metabolite 11-dehydro-TXB2 and either body mass and plasma fasting or postprandial glucose. The exact role of adiposity, adipose tissue inflammation, insulin resistance, and hyperglycemia in persistent platelet hyperreactivity in diabetes is difficult to clarify. Hyperglycemia is not a strong risk factor for CVD
[117][118][235,236], as confirmed by the evidence that interventions aimed at reducing plasma glucose did not significantly reduce CV risk and mortality
[119][120][121][237,238,239]. Consistently, the pharmacological reduction in glycated hemoglobin (HbA1c) only modestly improved CVD risk and mortality
[117][121][235,239], whereas newer drugs, including GLP-1 agonists and gliflozins, beyond their glucose-lowering effects, have provided effective results in terms of reduction in CV risk
[122][123][124][240,241,242], thus indicating the need to modulate risk factors other than hyperglycemia to blunt atherothrombosis
[125][243].
Exercise Effects on Platelets in Diabetes. Besides cardio-pulmonary fitness and weight control, exercise training improves glycemic control and insulin-resistance in T2DM
[126][244] and is strongly recommended for its benefits on the CV system
[127][245]. A total of 106 randomized controlled trials involving 7438 patients were included in a recent meta-analysis aimed at evaluating exercise effects in adults with T2DM. In comparison with no exercise, low to moderate supervised aerobic/resistance exercise is associated with significant improvement of glycemic and lipid profile, body weight, and blood pressure
[61][187].
Regular physical exercise in diabetes also shows beneficial effects on platelet function (
Table 1). Specifically, aerobic training for 8 weeks determines a remarkable reduction in MPV, PDW, and collagen-induced platelet aggregation
[128][246], attributable at least in part to downregulated glycoprotein (GP)IIb expression. A 12-week moderate-intensity aerobic exercise program was effective in upregulating platelets’ microRNAs (miRNA)-223 and downregulating P2RY12 receptor expression following decreased platelet aggregability in T2DM patients
[129][247], whereas short-term endurance training determined a positive impact on platelet function, glycemic indices, physical fitness, and body composition, but did not change miRNA-223 levels and P2RY12 expression
[130][248]. One year of exercise training was not effective in modifying platelet-derived microvesicles in T2DM patients with CAD, but decreased levels of PMVs carrying TF [CD61
+/CD142
+/Annexin V (AV)
+] and von Willebrand factor (vWF; CD31
+/CD42b
+/AV
+) in those with albuminuria
[131][249]. In another study, acute exercise increased platelet aggregation in diabetic subjects despite treatment with aspirin diabetics, thus showing the limited effects of aspirin in inhibiting exercise-induced platelet aggregation
[132][250]. The impaired action of aspirin could be partially explained by taking into account that endothelial dysfunction caused by inflammation and oxidative stress causes an impaired release of PGI2 and NO following acute exercise, thus limiting the antiplatelet effects of aspirin
[133][251]. Another randomized crossover design evaluated the short-term effects of post-meal walking exercise with and without a low-carbohydrate diet on vascular parameters. The authors found that a 15-min post-meal walk in addition to a diet significantly improved endothelial function, even if its role in platelet reactivity, as measured through PMP release and monocyte platelet aggregate (MPA) count and percentage, was unclear
[134][252]. The effects of postprandial hyperglycemia in impairing endothelial function and increasing oxidative stress are particularly concerning for their role in the excessive CVD risk in diabetes
[135][136][253,254], and the correction of hyperglycemia and oxidative stress can positively influence endothelial function, at least in an acute setting
[137][255]. A limitation of this study was certainly its short duration, which did not allow the observation of the endothelium-mediated benefits for platelets. In a study carried out by Scheinowitz et al., diabetic patients in antiaggregating therapy with aspirin were enrolled and undertook acute exercise. Platelet samples at rest and immediately post-exercise were stimulated with agonists, and the expression of the pan-platelet marker CD41 and platelet activation marker CD62P was measured
[138][256]. Despite diabetic patients showing systolic blood pressure significantly higher than non-diabetics, no differences were found in platelet parameters. Finally, platelet CD markers of platelet activation did not change in a study comparing the effects of blood-flow restriction under low-intensity resistance exercise (20%) versus high-intensity resistance exercise (80%) in female T2DM patients, even though CD62P, CD61, CD41, and CD42 were reduced following resistance exercise in both trials independently of blood-flow restriction conditions
[139][257] (
Table 1).
Table 1. Exercise and platelet parameters in metabolic diseases. Abbreviations: adenosine diphosphate (ADP); adenosine triphosphate (ATP); thromboxane B2 (TXB2); platelet (PLT); coronary artery disease (CAD); cardiac rehab (CR); high caloric CR (HCR); extracellular vesicles (EVs); exercise (EX); high carbohydrate and fat diet (HCFD); high-fat (HF); high-fat + exercise (FE); oxidized Low-Density Lipoprotein (ox-LDL); prostacyclin (PGI2); coronary heart disease (CHD); high-density lipoprotein cholesterol (HDL-c); prostaglandin F2α (PGF2α); mean platelet volume (MPV); platelet distribution width (PDW); plateletcrit (PCT); type 2 diabetes mellitus (T2DM); monocyte-platelet aggregates (MPAs); blood-flow restriction (BFR).