1. Oxidative Sstress in Hhealthy Red Blood Cred blood cell
Red blood cells (RBCs) are exposed to endogenous and exogenous oxidants, commonly referred to as reactive oxygen species (ROS) and reactive nitrogen species (RNS). These oxidants comprise a large group of molecules with different properties, including cellular sources and preferred molecular targets, which and have been discussed in more detail elsewhere [1, [1][2]2].
One of the main mechanisms of endogenous oxidant production involves oxyhemoglobin (HbO2). The autoxidation of HbO2 occurs spontaneously at a low rate, to yield superoxide (O2•–) and methemoglobin (Hb-FIII, MetHb) [3]. Superoxide itself is a weak oxidant, but can further react to make stronger oxidants. Superoxide can spontaneously dismutate to yield hydrogen peroxide (H2O2) and oxygen [4]. Hydrogen peroxide is a stronger oxidant that will react with thiols and metal centers [5]. Hydrogen peroxide reacts with HbO2 to yield ferryl hemoglobin, which can oxidize other proteins and lipids [6]. Furthermore, in the presence of one electron reductants such as FeII, H2O2 can also generate hydroxyl radical (HO•), one of the strongest biological oxidants [4]. Hydroxyl radical will react with most organic molecules at diffusion-controlled rates, to yield organic radicals that can propagate the oxidative damage [6][7].
RBCs will also be exposed to oxidants derived from endothelial and immune system cells, which generate nitric oxide (NO•), superoxide, peroxynitrite (ONOO-), H2O2 and hypochlorous acid (HOCl). Nitric oxide is produced by the endothelial enzyme nitric oxide synthase (NOS3) as a signal molecule to induce vasodilation, and by immune system cells at larger amounts by inducible NOS2, that result in the formation of more potent oxidizing species that can kill invading pathogens [7].[8]
Proteins and lipids can be damaged by oxidants in RBCs. The ultimate result of oxidative damage to RBCs is hemolysis, the loss of membrane integrity and the release of hemoglobin and the other intracellular proteins. Free hemoglobin is particularly toxic and this is evident in several RBCs diseases [8][23].
The membrane of RBCs is composed of phospholipids, cholesterol, glycolipids and proteins (some of them glycosylated). The polyunsaturated fatty acids (PUFA), 18% of the total fatty acids in RBC [9][24], are the lipid components that are more susceptible to oxidation, in a series of reactions that trigger lipoperoxidation. The first event is the abstraction of a bis-allylic hydrogen from a polyunsaturated fatty acid, to yield a lipid derived radical, which rapidly reacts with molecular oxygen to yield a lipid-derived peroxyl radical (LOO•). This LOO• can subsequently subtract a hydrogen from a neighboring PUFA, and the reaction propagates as a chain reaction [10][25]. The further oxidation of LOOH yields reactive aldehydes such as hydroxynonenal and malondialdehyde.
Although the RBCs are exposed to large amounts of oxidants, both from endogenous and exogenous sources, they are well prepared to resist. Robust antioxidant defenses allow normal RBCs to survive 120 days in circulation. The main defenses against oxidant damage are provided by different enzymatic systems, aided by low molecular weight antioxidant and electron-rich molecules. The antioxidant defenses ultimately rely on the reducing power of NADPH, obtained from the oxidation of glucose by the pentose phosphate pathway.
As developed above, RBCs oxidative stress strongly depends on the balance among pathophysiological mechanisms producing ROS and enzymatic and non-enzymatic antioxidant systems. In this second part of the resviearch, researchersw, we aim to explore several RBC diseases where this balance is strongly altered both by increased ROS production or by diminished antioxidant capacity. This selection of RBC diseases is not exhaustive.
2. Sickle Cell Disease
Sickle cell disease (SCD) is an inherited genetic disorder resulting in the production of an abnormal hemoglobin S (HbS) that undergoes deoxygenation-dependent polymerization
[11][12][122,123]. The repeated cycles of HbS polymerization induce RBCs’ shape distortion, cell rigidity, cell membrane alteration, and fragility, ultimately resulting in intravascular and extravascular hemolysis. Behind this primary described pathological mechanism, SCD pathophysiology appears to be more complex and involves an intricate network of molecular and cellular partners. In fact, in addition to HbS polymerization, an imbalance of the redox status is also observed in SCD due to an increase in the production of ROS and/or RNS conjugated to an impairment of the antioxidant systems (
Figure 1 SCD panel). For example, peroxynitrite is involved in the oxidation and nitration of several intracellular targets (thiols, protein-membrane, lipids), leading to breakage of DNA, impairment of cell signaling and cell death (reviewed in
[13][14][124,125]). In SCD, oxidative stress can arise from sickle RBCs and/or activated neutrophiles, platelets, and endothelial cells (ECs). Several erythroid and non-erythroid mechanisms have been described accounting for this pro-oxidant environment: (i) HbS autooxidation, (ii) heme and iron release, (iii) increased NADPH oxidase and endothelial xanthine oxidase (XO) activity, (iv) decreased NO
• bioavailability, (v) erythroid mitochondrial retention
[15][126].
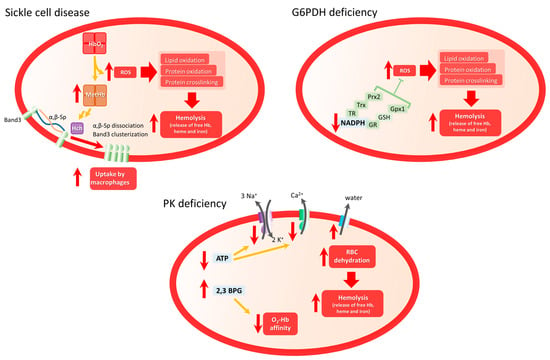
Figure 1. Scheme of main mechanism involved in oxidative stress and hemolytic clinical manifestations in Sickle Cell Disease, G6PDH deficiency, and PK deficiency. In SCD, highly unstable HbS will be converted in MetHb, favoring band 3 clustering and dissociation from membrane complexes, inducing membrane disorganization and membrane fragility. In G6PD, dramatic reduction of NADPH levels diminishes the antioxidant capacity of RBCs increasing ROS-induced hemolysis. In PKD, diminished ATP levels affect the functioning of membrane proteins such as Na+/K+ pump or PMCA pump, which will indirectly induce water efflux and RBC dehydration, incrementing RBC fragility and hemolysis.
HbS is very unstable and could easily undergo autoxidation in the presence of oxygen. The reaction leads to the production of MetHb that no longer binds oxygen and O
2•− that dismutates to H
2O
2 [16][17][18][19][127,128,129,130]. This results in oxidative damage of the RBC membrane and lipid and protein oxidation, leading to hemolysis and release of toxic heme leading to the exacerbation of the pro-oxidative environment (see below). HbS-induced oxidative stress leads to post-translational modifications of hemoglobin (notably oxidation of Cys93 and ubiquitination of Lys96 and Lys145 of the β globin), phosphorylation of band 3, the most abundant protein of the RBC membrane, and ubiquitination of other erythroid proteins. ROS-induced band 3 modification induces its clusterization and dissociation from membrane/cytoskeleton complexes, leading to RBC membrane disorganization and potentially microparticle formation
[20][21][22][131,132,133]. Several studies have highlighted the role of microparticles in several SCD complications, such as vaso-occlusion and kidney dysfunction
[23][24][25][134,135,136].
Repeated cycles of sickling/unsickling lead to the fragilization of the RBC membrane and thus to hemolysis that results in the release of extracellular hemoglobin, free heme, and free iron, all highly toxic to the vasculature by triggering vascular oxidative burden
[8][26][27][22,137,138]. In fact, oxidative stress generated at the erythroid levels can affect not only RBCs but also neutrophils, monocytes, and endothelial cells. The released heme and ATP from hemolyzed RBCs will act as damage-associated molecular patterns (DAMPs), promoting the activation of endothelial cells, macrophages, and neutrophils through different cellular pathways involving several receptors such as P2X7, toll-like receptor 4 (TLR4) or other unidentified receptors. Those activation processes trigger the expression of adhesion molecules at the cell surface and also pro-inflammatory mediators resulting in the exacerbation of the pro-inflammatory and oxidant environment. These can ultimately lead to vaso-occlusion and other SCD complications. Heme promotes adhesion events and thus vaso-occlusion through the von Willebrand factor (vWF) release from endothelial granules, inter cellular adhesion molecule (ICAM-1), vascular cell adhesion molecule (VCAM-1), and P-selectin expression at the surface of the vessel wall. Thus, it promotes leukocyte recruitment to the vessel wall and leukocyte/sickle RBC interactions. In the SCD context, heme has also been shown to promote neutrophil activation and Neutrophil Extracellular Traps (NETs) that are composed of decondensed chromatin with cytoplasmic protein
[28][29][30][139,140,141]. Those NETs can, in turn, contribute to the activation of the vascular system through the activation of the TLR4/TLR9 signaling pathways, thus exacerbating the oxidative environment.
NADPH oxidase is one of the major enzymes responsible for the production of O
2•− in leukocytes, RBCs, and endothelial cells. ROS produced by erythroid NADPH contributes to the exacerbation of erythroid dysfunction by exacerbating cell stiffness resulting in the increase of hemolysis
[31][142]. Xanthine oxidase (XO) is also responsible for a large part of the production of O
2•− and H
2O
2. Its activity is increased in SCD plasma, but its source remains to be clearly identified
[32][143].
NO
• plays an important role in vascular homeostasis and physiology. Notably, it acts on smooth muscle cells by regulating the vascular tone as a vasodilator, and on endothelial cells through downregulation of the expression of members of the selectin family, such as ICAM-1 and VCAM-1
[33][144]. NO
• could also inhibit platelet activation
[34][145]. Interestingly, NO
• may also act on the RBC itself by modulating its deformability through, in part, the soluble guanylate cyclase (sGC)
[35][36][146,147].
In SCD, hemolysis and consecutive free extracellular hemoglobin release lead to NO
• scavenging, thus decreasing its bioavailability in the circulation. Furthermore, the production and release of O
2•− may participate in the decrease in NO
• through its reaction to form ONOO
− [37][148]. Therefore, the decreased NO
• bioavailability in SCD, negatively affects vascular tone regulation and expression of adhesion proteins.
Recently, Jagadeeswaran et al. showed that RBCs from SCD patients retain mitochondria
[38][149], and this was confirmed by other groups
[39][40][41][150,151,152]. However, the functionality of these mitochondria remains controversial. Some studies showed that they were still functional and that the mitochondrial retention was associated with high levels of ROS, but some of these observations have been made in SCD mice model or in a population of erythroid circulating cells that might also include reticulocytes, i.e., immature RBCs
[38][40][41][149,151,152]. Another group did not detect any activity of these retained mitochondria in mature RBCs
[39][150]. A clear link between mitochondrial retention and the increased oxidative stress in SCD remains to be fully determined as well as the mechanism leading to this mitochondrial retention.
It is well established now that oxidative stress plays an essential role in SCD pathophysiology and in complication occurrence. However, it appears that oxidative mechanisms are considerably complex as they involve not only the RBC as the primary pathological cell target but also vascular endothelial cells, monocytes, and neutrophils. The complexity is heightened by the intimate interplay between oxidative mechanisms and inflammation with the activation of innate immune cells and the production of pro-inflammatory mediators. A vicious circle sets in, exacerbating the pro-oxidative, pro-inflammatory, pro-coagulant, pro-adherent environment. This highly toxic milieu is deleterious in the short-term with the appearance of acute complications and also deleterious in the long-term, with end-organ damage. Consequently, new drugs targeting oxidative stress have been developed to counteract its detrimental consequences. To date, the main antioxidant therapy that has shown some benefits in SCD clinical trials is L-glutamine, an amino acid needed for the synthesis of nucleotides as NAD. Supplementing with L-glutamine could reduce the erythroid oxidative process and protect RBCs from oxidative damage. However, this treatment has shown limitations as some SCD patients did not tolerate the treatment, and it seems to fail to counteract anemia and hemolysis
[42][43][153,154]. This observation means that the mechanism underlying pro-oxidative stress in SCD requires a lot more investigation in order to identify new potential therapeutical targets.
32. Glucose 6-Phosphate Dehydrogenase Deficiency
G
lucose-6
-phosphate dehydrogenase (G6PDH)PDH catalyzes the first reaction in the pentose–phosphate pathway, oxidizing glucose-6-phosphate to 6-phosphogluconate and reducing NADP to NADPH, which is essential to provide reducing equivalents to several antioxidant systems
[44][45][46][155,156,157], as discussed above.
The G6PDH deficiency (G6PD) is a chromosome X-linked highly polymorphic genetic disorder characterized by the reduced activity of the enzyme. Although most G6PD patients do not normally present clinical manifestations, RBCs from these patients present lower levels of NADPH and are more susceptible to oxidative stress (
Figure 1 G6PD panel) induced by the action of drugs, anesthetics, infections, and metabolic disturbances
[46][47][48][49][157,158,159,160], leading to hemolytic anemia and various health complications (reviewed in
[45][46][156,157]).
G6PD is usually associated with favism, a hemolytic anemia syndrome induced by the ingestion of fava beans
[50][51][161,162]. However, even though patients presenting fava bean intolerance carry some G6PD polymorphism, not all G6PD patients are intolerant to fava beans. Actually, different metabolites from fava beans, such as vicine and divicine, are highly oxidant and could induce hemolysis by depleting the antioxidant capacity of RBCs in a mechanism similar to that of synthetic drugs
[52][53][54][163,164,165].
Interestingly, some of the drugs and compounds inducing hemolytic anemia in G6PD patients are not able to induce RBC hemolysis in vitro
[52][53][54][163,164,165], supporting the hypothesis of other genetic factors contributing to the hemolytic phenotype
[55][166]. Recently, Dinarelli et al. reported that RBCs from G6PD patients stored for 6–12 days were surprisingly less sensitive to hemolysis. Authors suggest that these aged RBCs presented a metabolic regulation leading to lower energy consumption and higher stress resistance
[56][167]; however, this hypothesis should be further confirmed by studies including a higher number of patients. Moreover, these results are contradictory with those of Francis et al., which show that, after 42 days of storage, the quality of post-transfusion RBCs is significantly lower in G6PD patients compared with control subjects
[57][168]. Infections, both from bacterial
[58][59][169,170] or viral
[60][61][62][171,172,173] origin, are also able to induce hemolytic anemia in G6PD patients, probably by inducing ROS production by circulating phagocytes.
Moreover, the severity of the clinical phenotype is patient-dependent. Looking for susceptibility factors, Tang et al. studied the metabolome changes in control or G6PD patients challenged by diamide-induced ROS production. They reported that diamide induced significant changes in RBC from G6PD patients leading to severe and irreversible loss of deformability
[63][174].
Finally, as G6PD alters several cellular processes under oxidative stress and is frequently associated with anemia, it could be expected a deficient RBC maturation. However, in vitro differentiation of CD34
+ hematopoietic progenitor cells isolated from patients with different G6PD severity did not show any alteration in progenitor proliferation, nor differentiation or enucleation
[64][175].
43. Pyruvate Kinase Deficiency
Pyruvate kinase (PK) is a critical enzyme in the glycolytic pathway, catalyzing the conversion of phosphoenolpyruvate (PEP) to pyruvate and generating ATP
[65][176]. Pyruvate kinase deficiency (PKD) is an autosomal (chromosome 1q21) recessive genetic disorder that affects RBCs’ ability to generate energy, leading to various degrees of hemolytic anemia
[66][67][68][69][177,178,179,180].
Lacking mitochondria, mature RBCs’ ATP production depends exclusively on glycolysis. Thus, impaired or reduced PK activity in PKD patients leads to a dramatic decrease in ATP levels, which are necessary for maintaining the cell’s integrity and deformability
[70][71][181,182]. Indeed, the main RBC membrane pumps controlling calcium, sodium, and potassium transport across the RBC membrane are P-type ATPase pumps, whose activity depends on ATP concentration. Reduced activity of these pumps leads to an altered ion balance that then induces water leak leading to RBC dehydration
[72][183] (
Figure 1 PKD panel). As a consequence, RBCs from PKD patients present altered membrane properties and become more susceptible to premature destruction at the spleen, leading to hemolytic anemia
[73][184]. Other than the elimination of altered mature RBCs, PKD patients also present a diminished number of reticulocytes, which are most susceptible to low ATP levels.
Another consequence of PK deficiency is the accumulation of glycolytic intermediates such as 2,3-biphosphoglycerate (2,3-BPG), which diminishes the O
2-hemoglobin affinity favoring the tissue oxygenation that could partially compensate for anemia
[74][75][185,186].
In a recent elegant article, Roy et al. developed a metabolomic approach to characterize some changes in metabolism pathways from PKD patients. They demonstrated that RBCs from PKD patients present higher levels of oxidative stress markers, such as polyamines, sulfur-containing compounds, and deaminated purines, correlated with increased pentose phosphate pathways metabolites. Moreover, these patients also showed higher levels of poly- and highly-unsaturated fatty acids and acyl carnitine
[76][187].