Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Yulin Feng.
Proteins have been extensively studied for their outstanding functional properties, while polyphenols have been shown to possess biological activities such as antioxidant properties. There is increasing clarity about the enhanced functional properties as well as the potential application prospects for the polyphenol–protein complexes with antioxidant properties. It is both a means of protein modification to provide enhanced antioxidant capacity and a way to deliver or protect polyphenols from degradation.
- complex
- interaction
- antioxidation
- free radical scavenging
1. Introduction
Protein’s excellent functional properties make it an important ingredient of food engineering a foaming agent, emulsion, film, gel, etc. [1]. It is also focused on improving its functional characteristics through appropriate structural modification [2]. Polyphenols (including flavonoids, phenolic acids, tannins, stilbene, curcumin, Figure 1) have excellent biological activity such as antioxidant capacity and are therefore expected to be more widely used in food systems. However, the low stability, degradability, and susceptibility to oxidation limit their application [3]. In recent years, the complexes formed by proteins (such as whey protein, egg protein, soy protein, zein, gelatin, casein) combined with polyphenols (including flavonoids, phenolic acids, tannins, stilbene, lignin, curcumin) has become both an effective mean of protein modification or processing with expanded functions and a way to protect or deliver polyphenols with biological activities from degradation, therefore, have shown increased attention and research heat [4,5,6,7,8,9,10][4][5][6][7][8][9][10].
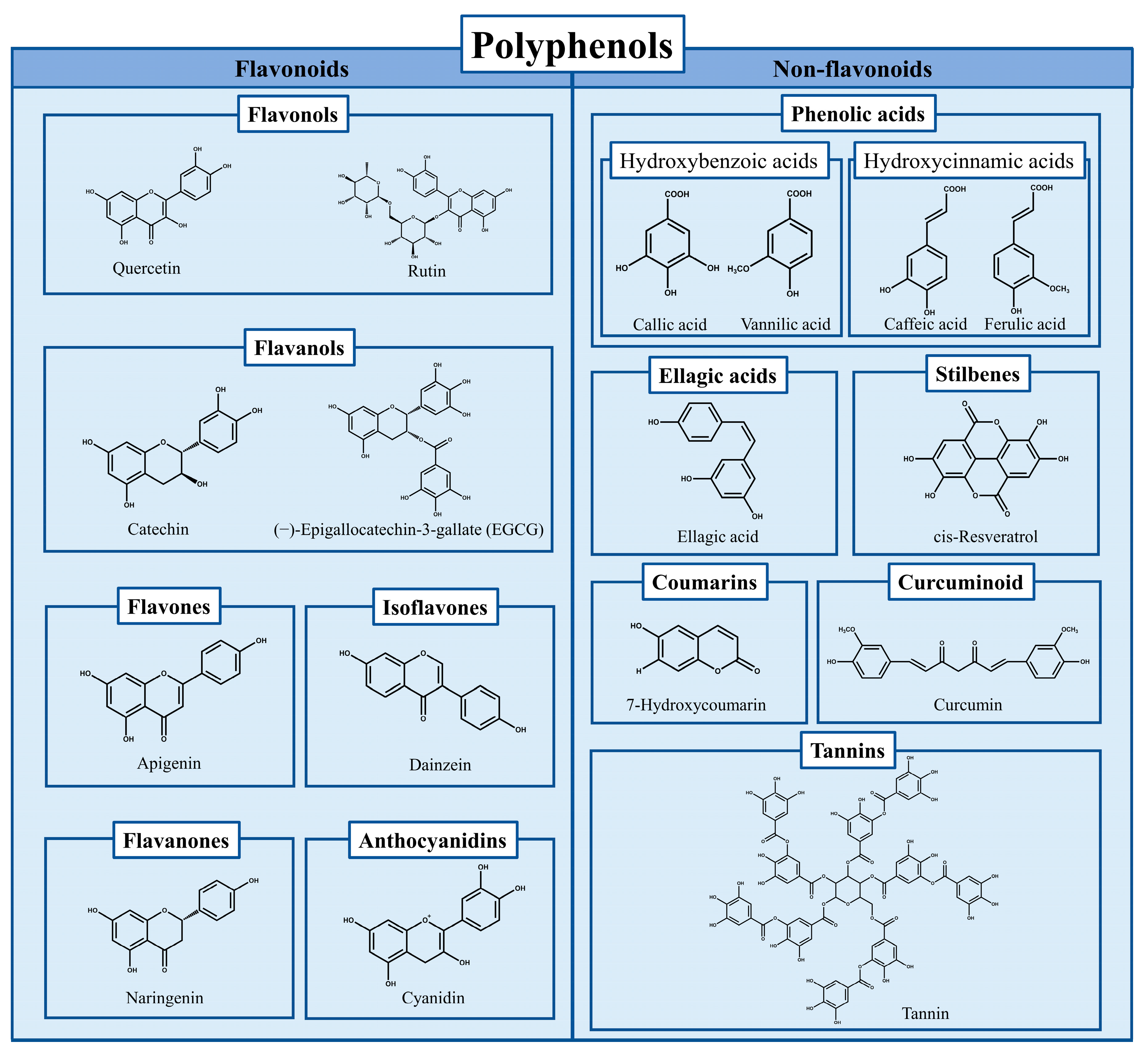
Figure 1. The classification of polyphenols and the chemical formulas of representative compounds.
Polyphenols can bind to proteins either covalently or non-covalently [5,7][5][7]. The enhanced antioxidant activity and functional properties of the complexes, such as emulsifying, gelling, and stability, are being widely studied [7]. Among them, antioxidant activity is one of the most essential characteristics of polyphenol–protein complexes. On the one hand, combining the proteins and polyphenols can introduce the active hydroxyl group from polyphenols into proteins so that it can endow proteins with significantly increased antioxidant activity [11,12,13][11][12][13]. This makes the polyphenol–protein complexes used as antioxidant emulsifiers, antioxidant films, etc. [7]. The emulsion prepared by Ren et al. [14] using the covalent combination of zein and resveratrol had higher antioxidant activity. The food packaging films prepared by Jiang et al. [15] applying the interaction between proteins and polyphenols showed high free radical scavenging activities. On the other hand, the combination with proteins can protect polyphenols from degradation and has better antioxidant stability [3]. Zou et al. [16] reported that the antioxidant activity of grape seed procyanidins could be protected from reducing activity loss during storage by soy protein isolates. The resveratrol encapsulated with nano-delivery particles constructed by Fan et al. [17] by conjugating bovine serum albumin (BSA) and caffeic acid had better digestive stability and cell antioxidant capacity. Furthermore, polyphenols and proteins can sometimes work synergistically to counteract oxidative [18].
2. Interaction Mechanism and Preparation of Polyphenol–Protein Complexes
In general, polyphenols may bind with proteins via covalent or non-covalent interactions [19] (Figure 2). An example of non-covalent interaction is hydrogen bonding, hydrophobic forces, electrostatic interactions, and van der Waals forces. In comparison, the covalent binding of polyphenols and proteins can be obtained primarily by enzymatic, alkaline, radical grafting, or chemical coupling methods [20].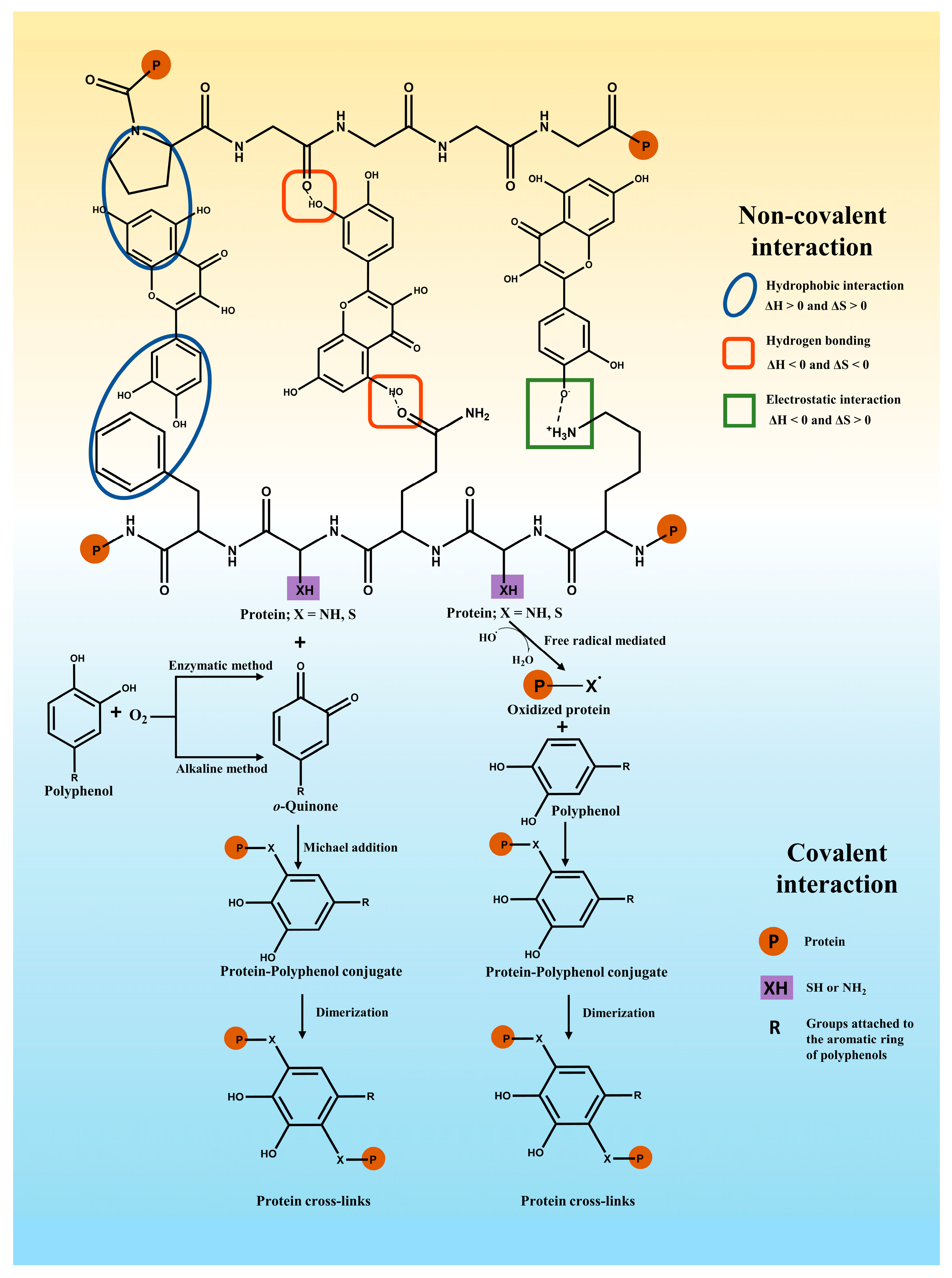
Figure 2. Major non-covalent interaction forces (the top half) between polyphenols and proteins and covalent bonding of polyphenols to proteins (the bottom half).
2.1. Non-Covalent Interactions
Non-covalent interactions between proteins and polyphenols may typically include hydrophobic interactions, hydrogen bonds, electrostatic interactions, and van der Waals interactions, which are reversible and weaker than covalent interactions [3,21][3][21]. In general, the formation of polyphenol–protein complexes relies mainly on hydrogen bonding and hydrophobic interactions, followed by other interactions (e.g., electrostatic interactions) [7]. When proteins and polyphenols interact with each other, alterations in the total strength of molecular interactions cause changes in the heat of the system. Information about the combined thermodynamic parameters (ΔH, ΔS, and ΔG) has been used to determine the nature of the interaction forces involved [22]. The main non-covalent interactions for ΔH > 0 and ΔS > 0 involve hydrophobic interactions. The main non-covalent interactions for ΔH < 0 and ΔS < 0 involve hydrogen bonding and van der Waals interactions. ΔH < 0 and ΔS > 0 interactions have been mainly attributed to electrostatic interactions [5,22][5][22]. Hydrogen bonding interactions are one of the main drivers of polyphenols binding to proteins. Hydrogen bonding is the interaction involving a hydrogen atom located between a pair of other atoms having a high affinity for electrons. As for polyphenols, they act as hydrogen donors, and their hydroxyl groups can form hydrogen bonds through interactions between the C=O groups of the amide group on the peptide chain, the oxygen or nitrogen on the side chains of amino acid residues, especially hydroxyl (–OH) and amino (–NH2) groups [23,24][23][24]. Zhang et al. [25] found that ferulic acid, quercetin, and vanillic acid could interact via three, seven, and two hydrogen bonds with β-lactoglobulin, respectively. Wen et al. [26] revealed that ovalbumin and procyanidin have a hydrogen bond-dominated interaction. Jiang et al. proved that Trp 118, Glu 11, and Lys 5 of α-lactalbumin could form hydrogen bonds with hydroxy safflower yellow A, respectively [11]. In addition to hydrogen bonding, hydrophobic interactions are one of the main driving forces of polyphenol–protein binding [3,6][3][6]. Hydrophobic interaction is usually understood as the force that the hydrophobic groups cluster together to avoid contact with water. The hydrophobic interactions rely on the fact that the non-polar aromatic ring in the phenolic compounds interacts hydrophobically with the hydrophobic amino acid residues of proteins (alanine, cysteine, glycine, isoleucine, leucine, methionine, phenylalanine, tyrosine, tryptophan, and valine) [5,27][5][27]. For instance, the aromatic ring and aliphatic chain of curcumin could interact hydrophobically with the hydrophobic region (residues 971-1410) of myosin [28]. Rosmarinic acid was inserted into the hydrophobic pocket formed by the amino acid residues Ser191, Arg198, Leu237, His241, Leu259, Ile263, His287, Ala290, and Glu291 of BSA and bound to the hydrophobic amino acids in the lumen of BSA through hydrophobic interactions [29]. The catechol part of chlorogenic acid interacted via the PI-PI accumulation of hydrophobic force with β-lactoglobulin Phe105 [30]. Electrostatic interactions occur as an attraction force that is created between two completely or partially ionized species with opposite charges. Electrostatic interactions between proteins and phenolics usually involve the deprotonation of some phenolic acids with low pKa values (e.g., cinnamic acid derivatives such as ferulic acid) under neutral conditions. At this point, positively charged protein groups, such as the ε-amino group of lysine, would react with the hydroxyl groups with a high electronegativity of the polyphenol [3,23][3][23]. For instance, ferulic acid has been found to interact electrostatically with sites containing positively charged amino acid residues in BSA since ferulic acid (pKa = 4.58) is a negatively charged molecule at pH 7.4 [31]. Similarly, the electrostatic interactions are more important for the binding of caffeic acid, ferulic acid, and chlorogenic acid (pKa = 3.45, 3.58, and 3.33, respectively) to β-casein since they are more readily ionized in aqueous solution under physiological pH conditions [32]. Van der Waals forces relatively attract the weak forces between molecules arising from electron fluctuations and the interaction of dipole moments. Van der Waals forces are usually generated in combination with other interactions [3,7][3][7]. Rosmarinic acids interacted with β-lactoglobulin or α-lactalbumin with the driving forces of hydrogen bonds, hydrophobic forces, and van der Waals force [29]. The binding of ferulic acid/quercetin/vanillic acid to β-lactoglobulin involved various non-covalent interactions such as hydrogen bonds, van der Waals interactions, and hydrophobic interactions [25]. Ovalbumin-isoquercitrin complexes are formed by hydrophobic interactions, van der Waals forces, and hydrogen bonding [33].2.2. Covalent Interactions
When covalent binding between polyphenols and proteins occurs, it is an irreversible interaction because of the chemical reactions involved [34]. The process mainly concerns the oxidation of polyphenols to strongly electrophilic quinones. Then, the interaction of the quinones with nucleophilic amino acid (cysteine, lysine, methionine, and tryptophan) residues on proteins or peptides via Michael addition forms covalent cross-linking [35]. Quinones can interact with sulfhydryl, amino, guanidinium, or imidazole groups on proteins or peptides [35], where free sulfhydryl groups have been identified as being more susceptible to covalent cross-linking than other groups [36]. Enzymatic and non-enzymatic methods are applied to mediate the covalent binding of polyphenols to proteins [6,37][6][37]. Enzymatic methods are environmentally friendly and highly specific methods that synthesize complexes with intense free radical scavenging activity. However, the preparation procedures are complex and expensive [38]. In this scheme, firstly, phenolase (monophenolase or cresolase) induces the oxidation of polyphenols to o-diphenols. Subsequently, under oxygen conditions, o-diphenolase (laccase or catecholase) converts the o-diphenols to o-quinones, and the active quinones can interact with nucleophilic amino acid residues in the protein chain to form cross-linked proteins or polymers [39,40][39][40]. For instance, Temdee and Benjakul [41] used laccase oxidized gallic acid and catechuic acid covalently cross-linked with gelatin from cuttlefish (Sepia pharaonis) skin to improve its gel functional properties. Velickovic and Stanic-Vucinic [39] et al. used tyrosinase and laccase to achieve covalent binding of caffeic acid to β-casein or β-lactoglobulin, and the reduced solubility and in vitro digestibility of the complexes were found. The alkaline reaction is one of the common non-enzymatic methods for binding polyphenols and proteins. The oxidation of polyphenols leads to the formation of semiquinones, which are rearranged into quinones under alkaline and aerobic conditions. These intermediates can form covalent crosslinks between proteins and polyphenols (C-N or C-S) [7,20,40][7][20][40]. Parolia et al. [18] obtained the conjugate of lentil protein and quercetin/rutin /ellagic acid prepared by the alkaline reaction, and the enhanced antioxidant properties of the conjugates were observed. Xu et al. [42] formed the conjugates by the binding of chlorogenic acid, gallic acid, and caffeic acid with zein under alkaline conditions and investigated the effects of covalent interactions on the structural and functional properties of the proteins. For non-enzymatic methods, free radical grafting with ascorbic acid and hydrogen peroxide as radical inducers is considered an effective synthetic method for the preparation of protein–polyphenol complexes with high bioactivity, low cost, and non-toxic chemicals involved [38,43][38][43]. The process mainly involves the oxidation of amino acids located on the side chains of proteins by free radical initiators to form free radicals, which then react with polyphenols through covalent bonds to form polyphenol–protein conjugates with strong interactions as well as high stability [44,45][44][45]. The wheat gluten hydrolysate-chlorogenic acid conjugate was obtained via the free radical method to explore the potential application in improved functional properties of the conjugates [46]. A camel whey–quercetin conjugate was prepared using the redox pair consisting of ascorbic acid and H2O2 by Baba et al. [47]. In addition to the above methods, some chemical cross-linking agents were also applied to prepare polyphenol–protein conjugates [20]. The conjugates prepared from zein assembled with chlorogenic acid or gallic acid via the chemical method obtained by Xu et al. [48] had high polyphenol content and grafting efficiency. 1-ethyl-3-(30-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were applied to achieve caffeic acid binding with β-lactoglobulin for the development of adding functionality to milk-based protein [49]. According to the mechanism of interaction between polyphenol and protein presented above, the polyphenol–protein complexes can be prepared by physical mixing (non-covalent interaction), enzyme reaction, alkaline reaction, and free radical grafting (Figure 3). In general, different combination modes will be selected according to the various research purpose or the functional characteristics and biological activities intending to be improved. On the one hand, the preparation of non-covalent polyphenol–protein complexes (i.e., simple physical mixing under appropriate extrinsic conditions) is more conveniently prepared than covalently combined complexes [4]. Since the non-covalent interaction of the complexes is reversible, it is possible to achieve the binding of polyphenols to proteins during preparation and release of polyphenols during digestion [3,37][3][37]. Therefore, this combination mode is generally used to explore the digestion or release characteristics of the combined polyphenols, especially the protective effect of protein on antioxidant or other bioactive properties of polyphenols in the process of storage, intake, or digestion [12,30,50][12][30][50]. On the other hand, the covalent interaction between polyphenol–protein complexes is irreversible, which makes the complexes more stable. At the same time, covalent binding can mediate the high grafting rate of polyphenol binding to proteins so that polyphenol–protein complexes with higher antioxidant properties can be obtained [38,42,48][38][42][48]. Therefore, the complexes of covalent graft complexes are mainly used to prepare protein-based emulsifiers, delivery carriers, etc. Notably, the safety and potential hazard of the generated “new complexes” should also be considered, especially the conjugated complexes prepared via the irreversible chemical reaction. Therefore, researchers should consider the investigation purpose and application aspects to select the binding mode of polyphenol–protein complexes.
Figure 3. Production paths of protein–polyphenol complexes.
3. Assessment of Antioxidant Properties of Polyphenol–Protein Complexes
The methods used to assess antioxidants are abundant, and here is a summary and review of the methods commonly used to evaluate the antioxidant properties of polyphenol–protein complexes. The various assessment methods and their principles are summarized in Figure 4.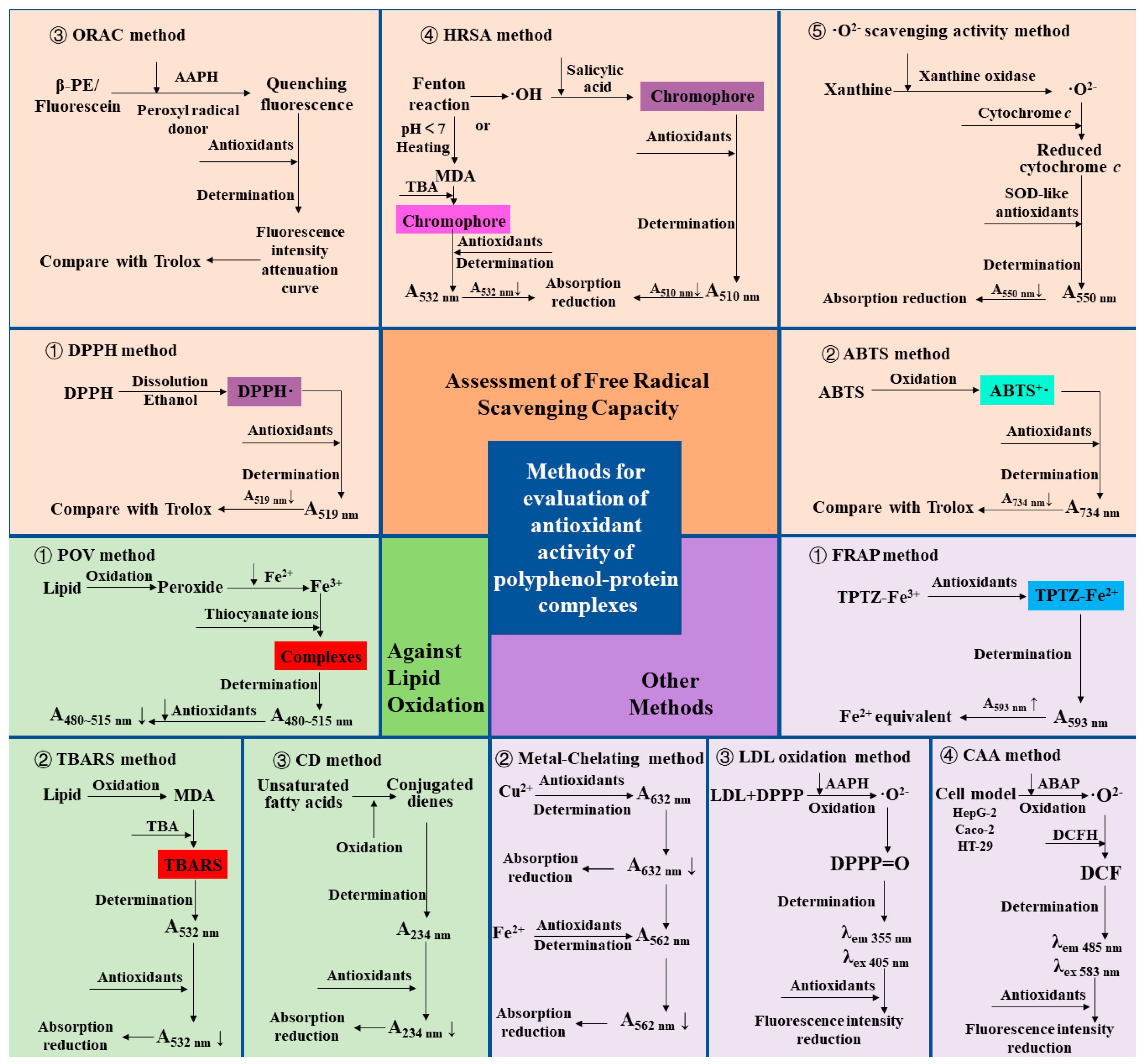
Figure 4. Assessments and principles of Antioxidant Capacity of polyphenol–protein complex. DPPH: 2,2-diphenyl-1-picrylhydrazyl radical; ABTS: 2,2′-azinobis (3-ethylbenzthiazoline-6-sulfonic acid) radical; ORAC: Oxygen radical absorbance capacity; β-PE: β-phycoerythrin; AAPH: 2,2′-Azobis (2-amidopropane) dihydrochloride; HRSA: Hydroxyl radical scavenging activity; MDA: Malondialdehyde; TBA: Thiobarbituric acid; ·O2−: Superoxide anion radical; SOD: Superoxide dismutase; FRAP: Ferric ion reducing antioxidant power; TPTZ-Fe3+: Tripyridine triazine ferric; TPTZ-Fe2+: Tripyridine triazine ferrous; LDL: Low-density lipoprotein; DPPP: Diphenyl-1-pyrenylphosphine; AAPH: 2,2’- azobis (2-amidinopropane) dihydrochloride; DPPP = O: Diphenyl-1-pyrenyl phosphine oxide; CAA: Cellular antioxidant activity; ABAP: 2,2′-azobis(2-amidinopropane) dihydrochloride; DCFH: Dichlorofluorescin probe; DCF: Dichlorofluorescein; POV: Peroxide value; TBARS: Thiobarbituric acid reactive substances; CD: Conjugated dienes.
3.1. Assessment of Free Radical Scavenging Capacity
The method to determine the antioxidant capacity of the complexes by assessment of free radical scavenging capacity is the most popular and used method at present because it is simple and fast. Among them, assessments of 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical and 2,2′-azinobis (3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) radical scavenging capacities are the most widely used methods. DPPH· ethanol solution is purple and has strong ultraviolet absorption at 517 nm. After adding antioxidants, the antioxidant reacts with DPPH·, making the reaction system lighter in color, and the absorbance value decreases [74][51]. In the ABTS method, ABTS, as the chromogenic agent, produces a stable blue-green cationic radical ABTS+· after oxidation, and then the reaction system is discolored by adding antioxidants. The absorbance was measured at 734 nm, and a decrease in absorbance was observed [75][52]. The results of the DPPH or ABTS radical scavenging capacity of the antioxidants were mostly expressed in terms of the equivalent concentration of Trolox [76][53]. It is reported that the combination of chlorogenic acid and the proteins (zein, β-lactoglobulin, wheat gluten hydrolysate) showed enhanced capacities of scavenging DPPH and ABTS free radicals [30,42,46,48][30][42][46][48]. In addition to DPPH and ABTS methods, the oxygen radical absorbance capacity (ORAC) method, hydroxyl radical scavenging activity (HRSA) method, and superoxide anion radical (·O2−) scavenging activity method is also used to evaluate the free radical scavenging capacity of polyphenol–protein complexes. In the ORAC method, β-phycoerythrin (β-PE) or fluorescein (3′,6′-dihydroxyspiro[isobenzofuran-1[3H], 9′[9H]-xanthen]-3-one) are used as the fluorescent indicator protein, and the 2,2′-Azobis (2-amidopropane) dihydrochloride (AAPH) and Cu2+-H2O2 system are used as the sources of lipid peroxidation free radicals or hydroxyl free radicals, with the Trolox as the reference in general. When β-PE is attacked by free radicals, the fluorescence decreases continuously at a certain wavelength. The free radical scavenging ability of the sample is calculated according to the change in its fluorescence intensity attenuation curve [77,78][54][55]. In the HRSA method, one is to generate hydroxyl radicals to initiate the Fenton reaction. The oxidation degree of the system is evaluated by measuring the amount of Fenton reaction products. The other is evaluated by measuring the amount of hydroxyl radicals produced by the Fenton reaction. In the former, deoxyribose, iron, and EDTA produce hydroxyl radicals to induce the Fenton reaction. When heated under acidic conditions, malondialdehyde (MDA) is produced, and thiobarbituric acid (TBA) will interact to form a pink chromophore with absorption at 532 nm. The degree of oxidation of the system is evaluated by measuring the absorbance at 532 nm. In the latter, H2O2 /Fe2+ system generates hydroxyl radicals through the Fenton reaction and produces purple compounds with salicylic acid addition. After adding antioxidants, the reduction of absorption at 510 nm was measured to reflect its hydroxyl radical scavenging ability [79][56]. In ·O2− scavenging activity method, O2− produced in the xanthine/xanthine oxidase system can reduce a certain amount of oxidized cytochrome c to reduced cytochrome c, which has the maximum light absorption at 550 nm. In the presence of superoxide dismutase (SOD) or SOD-like antioxidants, due to their catalytic disproportionation of a part of O2−, the amount of reduced cytochrome c is correspondingly reduced, and the absorbance at 550 nm is reduced so as to evaluate the SOD-like O2− scavenging activity of antioxidants [65][57]. Fan et al. [57][58] prepared whey protein isolate-(−)-epigallocatechin-3-gallate (EGCG) conjugate by free radical grafting and used it as an emulsifier to stabilize menhaden oil. The results showed that the ORAC of the conjugate was enhanced compared with whey protein isolate alone. The results of the HRSA of Abd El-Maksoud et al. [49] showed that the covalently conjugate formed by β-lactoglobulin and chlorogenic acid has a more robust antioxidant capacity than the non-covalent complex. Chung et al. [65][57] found that gelatin did not have superoxide anion scavenging activity, while the enzyme synthesized gelatin catechin conjugate gave SOD-like activity.3.2. Other Assessment of Antioxidant Properties
The ferric ion-reducing antioxidant power (FRAP) method is widely used to determine the antioxidant activity indirectly (i.e., the ability of the tested substance to reduce ferric iron to ferrous iron) [80][59]. Yin et al. [60] found that after β-casein was combined with chlorogenic acid, the FRAP value of the complex was significantly higher than the sum of β-casein alone and chlorogenic acid alone, indicating a synergistic effect. Another report showed that the FRAP values of chlorogenic acid in the presence of β-lactoglobulin were significantly increased at high temperatures (85–121 °C) compared to the values of chlorogenic acid alone [61]. The antioxidant activity can also be evaluated by measuring the ability of antioxidants to chelate metal ions, which are one of the essential sources of free radicals [81][62]. It has been reported that the metal-chelating activity of silk sericin increased after modification by hydroquinone and pyrogallol, thus systematically reducing the degree of oxidation [64][63]. In the low-density lipoprotein (LDL) oxidation method, LDL is labeled by diphenyl-1-pyrenylphosphine (DPPP), a fluorescent probe that can reflect hydrogen peroxide produced by lipid oxidation [65][57]. Antioxidants can inhibit the oxidation of LDL, thereby reducing the fluorescence intensity. The method was applied to prove that the gelatin-catechin conjugate showed more potent inhibitory activity on LDL oxidation than unconjugated catechin [65][57]. To better reflect the effects of antioxidants in physiological conditions, the cellular antioxidant activity (CAA) method is created to evaluate the intracellular reaction of antioxidants to establish a better biological correlation with the bioavailability, absorption, and metabolism of antioxidants in cells. The pre-treated cells contained dichlorofluorescin (DCFH) probes. Through the action of 2,2′-azobis (2-amidinopropane) dihydrochloride (ABAP) on cells to generate peroxy radicals, DCFH is oxidized to dichlorofluorescein (DCF) with fluorescence, whose absorption and emission wavelengths are 485 nm and 583 nm respectively. Antioxidants can block the oxidation of DCFH to DCF. Therefore, the antioxidant capacity of antioxidants can be evaluated by reducing cell fluorescence [82][64]. Fan et al. [17] proved that resveratrol loaded by zein-BSA nanoparticles has higher antioxidant capacity than free state by using the CAA method, indicating that the nanoparticle delivery system improved the absorption and bioavailability of encapsulated antioxidant components into human colon carcinoma cell monolayers (Caco-2 cells). Hoskin et al. [66][65] found that blueberry polyphenol–protein particles maintained the cellular antioxidant activity of the blueberry extract in mouse macrophage RAW 264.7.3.3. Assessment of Capacities against Lipid Peroxidation
At present, the emulsifier or stabilizer used as a lipid system has become one of the crucial applications of polyphenol–protein complexes. For this purpose, in addition to the above methods for determining antioxidant activity, it is necessary to assess the antioxidant activity of the complex to inhibit the oxidation of the lipid system. Peroxide is the main primary product of the automatic oxidation of lipids [83][66]. The peroxide formed by lipid oxidation can oxidize Fe2+ to Fe3+ under acidic conditions. Then Fe3+ and thiocyanate ions form the red complex, which has the maximum absorption in 480~515 nm. By comparing the peroxide value (POV) of the lipid system with or without antioxidants in a storage time (during the process of lipid oxidation), the effect of antioxidants on inhibiting lipid oxidation can be obtained [84,85][67][68]. The second product of lipid oxidation is malondialdehyde (MDA). In the thiobarbituric acid reactive substances (TBARS) method, thiobarbituric acid (TBA) reacts with MDA under acidic conditions to form red compounds with absorption at 532 nm. Thus, the activity of inhibiting lipid oxidation can be evaluated by measuring the amount of MDA via the TBARS method in the lipid system containing antioxidants. By measuring the POV and TBARS value of the emulsion, Wen et al. [26] found that at 6-day, the oxidation degree of ovalbumin emulsion and ovalbumin–procyanidins complexes emulsion was 5.90% and 1.78%, respectively, reflecting that the addition of procyanidins improved the oxidation stability of the emulsion. The results of Zhao et al. [68][69] showed that the POV and TBARS values of the emulsion added with the conjugate of anchovy protein hydrolysate and phenols (catechin, gallic acid, and tannic acid) decreased significantly during storage time. The ability of the conjugate to inhibit oxidation was consistent with the trend of the antioxidant activity of polyphenols. In addition, the activity of maintaining the oxidative stability of the lipid system can be evaluated by measuring the number of conjugated dienes (CD) formed by unsaturated fatty acids in the lipid system [86][70]. Chen et al. [72][71] evaluated the antioxidant activity of the whey protein isolates-lotus seedpod proanthocyanin conjugate in the emulsion. The results showed that the conjugates had a lower CD value than whey protein isolates alone during the 15-day storage period, reflecting the higher antioxidant stability of the conjugates.3.4. Effects of Assessment Methods on Antioxidant Properties of the Complexes
As described above, various methods exist for evaluating the antioxidant properties of polyphenol–protein complexes. Researchers often use various ways to determine the antioxidant activity of the complexes. Generally, the determination results of different methods are consistent. However, due to the different principles of the methods, sometimes the results are inconsistent. Yin et al. [60] observed that the antioxidant activity of β-casein-chlorogenic acid complexes determined by FRAP and ABTS had opposite results. The results of FRAP showed that the complex has a synergistic effect. That is, the FRAP value of the complex was higher than the sum of β-casein and chlorogenic acid alone. On the contrary, ABTS free radical scavenging ability showed an antagonism effect of the complexes. Another study showed that although the complexes formed by whey protein isolate and EGCG, quercetin, apigenin, or naringenin, all showed higher antioxidant activity than natural whey protein isolate, the antioxidant capacity of the complexes combined with different polyphenols was observed in different orders in the ABTS and DPPH radical scavenging methods and the FRAP method [13]. It should be noted that most of the evaluation of the antioxidant activity of the complexes formed by the binding of polyphenols and proteins needs to be compared with natural proteins or phenolic acids alone to show the enhanced antioxidant activity of the complexes [13,61,87][13][61][72] or the masking effect of phenolic acids [11,50,62][11][50][73]. Sometimes, the antioxidant capacity of the complexes will be compared with the sum of the antioxidant activities of individual proteins and phenolic acids to judge the synergistic or antagonistic effect of the forming complexes in terms of antioxidant capacity [60,88,89][60][74][75]. The scholars also pointed out that although pure polyphenols showed more potent antioxidant activity than complexes, if the degree of combination or grafting rate of polyphenols in the complexes were considered in the calculation, it would be found that the combination of proteins and polyphenols synergistically enhanced their antioxidant activity [18]. To sum up, the assessment methods and comparison or calculation methods for evaluating the antioxidant capacity of polyphenol–protein complexes have an essential impact on the results. The choice of methods should take into consideration the experimental purpose or the characteristics of the composite, and the results of the determination should be compared. However, there are relatively few studies on the in vivo antioxidant properties of the complex, which should be given greater attention and more abundant studies in the future to demonstrate better the digestive properties, oral stability, bioaccessibility, and bioavailability of the complexes.References
- Tian, H.; Guo, G.; Fu, X.; Yao, Y.; Yuan, L.; Xiang, A. Fabrication, properties and applications of soy-protein-based materials: A review. Int. J. Biol. Macromol. 2018, 120, 475–490.
- Isaschar-Ovdat, S.; Fishman, A. Crosslinking of food proteins mediated by oxidative enzymes—A review. Trends Food Sci. Technol. 2018, 72, 134–143.
- Li, Y.; He, D.; Li, B.; Lund, M.N.; Xing, Y.; Wang, Y.; Li, F.; Cao, X.; Liu, Y.; Chen, X.; et al. Engineering polyphenols with biological functions via polyphenol-protein interactions as additives for functional foods. Trends Food Sci. Technol. 2021, 110, 470–482.
- Liu, J.; Yong, H.; Yao, X.; Hu, H.; Yun, D.; Xiao, L. Recent advances in phenolic-protein conjugates: Synthesis, characterization, biological activities and potential applications. RSC Adv. 2019, 9, 35825–35840.
- Baba, W.N.; McClements, D.J.; Maqsood, S. Whey protein-polyphenol conjugates and complexes: Production, characterization, and applications. Food Chem. 2021, 365, 130455.
- Li, M.; Ritzoulis, C.; Du, Q.; Liu, Y.; Ding, Y.; Liu, W.; Liu, J. Recent progress on protein-polyphenol complexes: Effect on stability and nutrients delivery of oil-in-water emulsion System. Front. Nutr. 2021, 8, 765589.
- Quan, T.H.; Benjakul, S.; Sae-leaw, T.; Balange, A.K.; Maqsood, S. Protein-polyphenol conjugates: Antioxidant property, functionalities and their applications. Trends Food Sci. Technol. 2019, 91, 507–517.
- Wang, X.; Zhao, Z. Improved encapsulation capacity of casein micelles with modified structure. J. Food Eng. 2022, 333, 111138.
- Iftikhar, M.; Zhang, H.; Iftikhar, A.; Raza, A.; Khan, M.; Sui, M.; Wang, J. Comparative assessment of functional properties, free and bound phenolic profile, antioxidant activity, and in vitro bioaccessibility of rye bran and its insoluble dietary fiber. J. Food Biochem. 2020, 44, e13388.
- Zhang, H.; Jin, C.; Lv, S.; Ren, F.; Wang, J. Study on electrospinning of wheat gluten: A review. Food Res. Int. 2023, 169, 112851.
- Jiang, Z.; Li, T.; Ma, L.; Chen, W.; Yu, H.; Abdul, Q.; Hou, J.; Tian, B. Comparison of interaction between three similar chalconoids and alpha-actalbumin: Impact on structure and functionality of alpha-lactalbumin. Food Res. Int. 2020, 131, 109006.
- Wei, Z.; Huang, Q. Impact of covalent or non-covalent bound epigallocatechin-3-gallate (EGCG) on assembly, physicochemical characteristics and digestion of ovotransferrin fibrils. Food Hydrocoll. 2020, 98, 105314.
- Liu, X.; Song, Q.; Li, X.; Chen, Y.; Liu, C.; Zhu, X.; Liu, J.; Granato, D.; Wang, Y.; Huang, J. Effects of different dietary polyphenols on conformational changes and functional properties of protein-polyphenol covalent complexes. Food Chem. 2021, 361, 130071.
- Ren, G.; Shi, J.; Huang, S.; Liu, C.; Ni, F.; He, Y.; Luo, X.; Li, T.; Song, Y.; Huang, M.; et al. The fabrication of novel zein and resveratrol covalent conjugates: Enhanced thermal stability, emulsifying and antioxidant properties. Food Chem. 2022, 374, 131612.
- Jiang, J.; Watowita, P.S.M.S.L.; Chen, R.; Shi, Y.; Geng, J.-T.; Takahashi, K.; Li, L.; Osako, K. Multilayer gelatin/myofibrillar films containing clove essential oil: Properties, protein-phenolic interactions, and migration of active compounds. Food Packag. Shelf Life 2022, 32, 100842.
- Zou, Y.C.; Wu, C.L.; Ma, C.-F.; He, S.; Brennan, C.S.; Yuan, Y. Interactions of grape seed procyanidins with soy protein isolate: Contributing antioxidant and stability properties. LWT-Food Sci. Technol. 2019, 115, 108465.
- Fan, Y.; Liu, Y.; Gao, L.; Zhang, Y.; Yi, J. Improved chemical stability and cellular antioxidant activity of resveratrol in zein nanoparticle with bovine serum albumin-caffeic acid conjugate. Food Chem. 2018, 261, 283–291.
- Parolia, S.; Maley, J.; Sammynaiken, R.; Green, R.; Nickerson, M.; Ghosh, S. Structure-Functionality of lentil protein-polyphenol conjugates. Food Chem. 2022, 367, 130603.
- You, J.; Luo, Y.; Wu, J. Conjugation of ovotransferrin with catechin shows improved antioxidant activity. J. Agric. Food Chem. 2014, 62, 2581–2587.
- Benjakul, S.; Singh, A.; Chotphruethipong, L.; Mittal, A. Protein-polyphenol conjugates: Preparation, functional properties, bioactivities and applications in foods and nutraceuticals. Adv. Food Nutr. Res. 2021, 98, 281–320.
- Jakobek, L. Interactions of polyphenols with carbohydrates, lipids and proteins. Food Chem. 2015, 175, 556–567.
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102.
- Buitimea-Cantua, N.E.; Gutierrez-Uribe, J.A.; Serna-Saldivar, S.O. Phenolic-protein interactions: Effects on food properties and health benefits. J. Med. Food 2018, 21, 188–198.
- Feng, Y.; Feng, X.; Liu, S.; Zhang, H.; Wang, J. Interaction mechanism between cereal phenolic acids and gluten protein: Protein structural changes and binding mode. J. Sci. Food Agric. 2022, 102, 7387–7396.
- Zhang, S.; Li, X.; Ai, B.; Zheng, L.; Zheng, X.; Yang, Y.; Xiao, D.; Sheng, Z. Binding of beta-lactoglobulin to three phenolics improves the stability of phenolics studied by multispectral analysis and molecular modeling. Food Chem. X 2022, 15, 100369.
- Wen, H.; Ning, Z.; Li, J.; Guan, Y.; Zhang, B.; Shang, X.; Liu, X.; Du, Z.; Liu, J.; Zhang, T. Stability of oil-in-water emulsions improved by ovalbumin-procyanidins mixture: A promising substrate with emulsifying and antioxidant activity. Colloids Surf. B Biointerfaces 2022, 215, 112473.
- Zembyla, M.; Murray, B.S.; Radford, S.J.; Sarkar, A. Water-in-oil Pickering emulsions stabilized by an interfacial complex of water-insoluble polyphenol crystals and protein. J. Colloid Interface Sci. 2019, 548, 88–99.
- Zhang, L.; Wang, P.; Yang, Z.; Du, F.; Li, Z.; Wu, C.; Fang, A.; Xu, X.; Zhou, G. Molecular dynamics simulation exploration of the interaction between curcumin and myosin combined with the results of spectroscopy techniques. Food Hydrocoll. 2020, 101, 105455.
- Lu, Y.; Zhao, R.; Wang, C.; Zhang, X.; Wang, C. Deciphering the non-covalent binding patterns of three whey proteins with rosmarinic acid by multi-spectroscopic, molecular docking and molecular dynamics simulation approaches. Food Hydrocoll. 2022, 132, 107895.
- Liu, J.; Song, G.; Yuan, Y.; Zhou, L.; Wang, D.; Yuan, T.; Li, L.; He, G.; Yang, Q.; Xiao, G.; et al. Ultrasound-assisted assembly of beta-lactoglobulin and chlorogenic acid for non covalent nanocomplex: Fabrication, characterization and potential biological function. Ultrason. Sonochem. 2022, 86, 106025.
- Ojha, H.; Mishra, K.; Hassan, M.I.; Chaudhury, N.K. Spectroscopic and isothermal titration calorimetry studies of binding interaction of ferulic acid with bovine serum albumin. Thermochim. Acta 2012, 548, 56–64.
- Li, T.; Li, X.; Dai, T.; Hu, P.; Niu, X.; Liu, C.; Chen, J. Binding mechanism and antioxidant capacity of selected phenolic acid—Beta-casein complexes. Food Res. Int. 2020, 129, 108802.
- Zhang, L.; Xu, L.; Tu, Z.C.; Wang, H.G.; Luo, J.; Ma, T.X. Mechanisms of isoquercitrin attenuates ovalbumin glycation: Investigation by spectroscopy, spectrometry and molecular docking. Food Chem. 2020, 309, 125667.
- Le Bourvellec, C.; Renard, C.M.G.C. Interactions between polyphenols and macromolecules: Quantification methods and mechanisms. Crit. Rev. Food Sci. Nutr. 2012, 52, 213–248.
- Loc Bao, P.; Wang, B.; Zisu, B.; Adhikari, B. Covalent modification of flaxseed protein isolate by phenolic compounds and the structure and functional properties of the adducts. Food Chem. 2019, 293, 463–471.
- Li, Y.; Jongberg, S.; Andersen, M.L.; Davies, M.J.; Lund, M.N. Quinone-induced protein modifications: Kinetic preference for reaction of 1,2-benzoquinones with thiol groups in proteins. Free Radic. Biol. Med. 2016, 97, 148–157.
- Wang, Y.; Xie, Y.; Wang, A.; Wang, J.; Wu, X.; Wu, Y.; Fu, Y.; Sun, H. Insights into interactions between food polyphenols and proteins: An updated overview. J. Food Process. Preserv. 2022, 46, e16597.
- Gu, L.; Peng, N.; Chang, C.; McClements, D.J.; Su, Y.; Yang, Y. Fabrication of surface-active antioxidant food biopolymers: Conjugation of catechin polymers to egg white proteins. Food Biophys. 2017, 12, 198–210.
- Velickovic, T.D.C.; Stanic-Vucinic, D.J. The Role of dietary phenolic compounds in protein digestion and processing technologies to improve their antinutritive properties. Compr. Rev. Food Sci. Food Saf. 2018, 17, 82–103.
- Liu, F.; Ma, C.; Gao, Y.; McClements, D.J. Food-grade covalent complexes and their application as nutraceutical delivery systems: A review. Compr. Rev. Food Sci. Food Saf. 2017, 16, 76–95.
- Temdee, W.; Benjakul, S. Effect of phenolic compounds and bark/wood extracts oxidised by laccase on properties of cuttlefish (Sepia pharaonis) skin gelatin gel. Int. Food Res. J. 2015, 22, 246–253.
- Xu, Y.; Wei, Z.; Xue, C.; Huang, Q. Covalent modification of zein with polyphenols: A feasible strategy to improve antioxidant activity and solubility. J. Food Sci. 2022, 87, 2965–2979.
- Feng, J.; Cai, H.; Wang, H.; Li, C.; Liu, S. Improved oxidative stability of fish oil emulsion by grafted ovalbumin-catechin conjugates. Food Chem. 2018, 241, 60–69.
- Liu, F.; Sun, C.; Yang, W.; Yuan, F.; Gao, Y. Structural characterization and functional evaluation of lactoferrin-polyphenol conjugates formed by free-radical graft copolymerization. RSC Adv. 2015, 5, 15641–15651.
- Spizzirri, U.G.; Iemma, F.; Puoci, F.; Cirillo, G.; Curcio, M.; Parisi, O.I.; Picci, N. Synthesis of antioxidant polymers by grafting of gallic acid and catechin on gelatin. Biomacromolecules 2009, 10, 1923–1930.
- He, D.; Xing, Y.; Wang, Y.; Zeng, W.; Gao, W.; Su, N.; Zhang, C.; Chen, H.; Xing, X.-H. Improved functional properties of wheat gluten hydrolysate by covalent conjugation with chlorogenic acid. Int. J. Food Sci. Technol. 2022, 58, 454–462.
- Baba, W.N.; Abdelrahman, R.; Maqsood, S. Conjoint application of ultrasonication and redox pair mediated free radical method enhances the functional and bioactive properties of camel whey-quercetin conjugates. Ultrason. Sonochemistry 2021, 79, 105784.
- Xu, Y.; Wei, Z.; Xue, C.; Huang, Q. Assembly of zein-polyphenol conjugates via carbodiimide method: Evaluation of physicochemical and functional properties. LWT-Food Sci. Technol. 2022, 154, 112708.
- Abd El-Maksoud, A.A.; Abd El-Ghany, I.H.; El-Beltagi, H.S.; Anankanbil, S.; Banerjee, C.; Petersen, S.V.; Perez, B.; Guo, Z. Adding functionality to milk-based protein: Preparation, and physicochemical characterization of beta-lactoglobulin-phenolic conjugates. Food Chem. 2018, 241, 281–289.
- Tong, X.; Cao, J.; Tian, T.; Lyu, B.; Miao, L.; Lian, Z.; Cui, W.; Liu, S.; Wang, H.; Jiang, L. Changes in structure, rheological property and antioxidant activity of soy protein isolate fibrils by ultrasound pretreatment and EGCG. Food Hydrocoll. 2022, 122, 107084.
- Tang, Y.Z.; Liu, Z.Q. Free-radical-scavenging effect of carbazole derivatives on DPPH and ABTS radicals. J. Am. Oil Chem. Soc. 2007, 84, 1095–1100.
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin. Sci. 1993, 84, 407–412.
- van den Berg, R.; Haenen, G.; van den Berg, H.; Bast, A. Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures. Food Chem. 1999, 66, 511–517.
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free Radic. Biol. Med. 1993, 14, 303–311.
- Ou, B.X.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626.
- Nicholas, S.; Quinton, J.C. Hydroxyl radical scavenging activity of compatible solutes. Phytochemistry 1989, 28, 1057–1060.
- Chung, J.E.; Kurisawa, M.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and antioxidant property of gelatin-catechin conjugates. Biotechnol. Lett. 2003, 25, 1993–1997.
- Fan, Y.; Liu, Y.; Gao, L.; Zhang, Y.; Yi, J. Oxidative stability and in vitro digestion of menhaden oil emulsions with whey protein: Effects of EGCG conjugation and interfacial cross-linking. Food Chem. 2018, 265, 200–207.
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76.
- Yin, Z.; Qie, X.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; Li, W.; He, Z. Effect of thermal treatment on the molecular-level interactions and antioxidant activities in beta-casein and chlorogenic acid complexes. Food Hydrocoll. 2022, 123, 107177.
- Qie, X.; Chen, W.; Zeng, M.; Wang, Z.; Chen, J.; Goff, H.D.; He, Z. Interaction between beta-lactoglobulin and chlorogenic acid and its effect on antioxidant activity and thermal stability. Food Hydrocoll. 2021, 121, 107059.
- Liu, Q.; Kong, B.; Xiong, Y.L.; Xia, X. Antioxidant activity and functional properties of porcine plasma protein hydrolysate as influenced by the degree of hydrolysis. Food Chem. 2010, 118, 403–410.
- Omar, A.; Arken, A.; Wali, A.; Gao, Y.; Aisa, H.A.; Yili, A. Effect of phenolic compound-protein covalent conjugation on the physicochemical, anti-inflammatory, and antioxidant activities of silk sericin. Process Biochem. 2022, 117, 101–109.
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907.
- Hoskin, R.T.; Xiong, J.; Esposito, D.A.; Lila, M.A. Blueberry polyphenol-protein food ingredients: The impact of spray drying on the in vitro antioxidant activity, anti-inflammatory markers, glucose metabolism and fibroblast migration. Food Chem. 2019, 280, 187–194.
- Lezerovich, A. Determination of peroxide value by conventional difference and difference-derivative spectrophotometry. J. Am. Oil Chem. Soc. 1985, 62, 1495–1500.
- Stine, C.M.; Harland, H.A.; Coulter, S.T.; Jenness, R. A Modified peroxide test for detection of lipid oxidation in dairy products. J. Dairy Sci. 1953, 37, 202–208.
- Lips, A.; Chapman, R.A.; Mcfarlane, W.D. The application of the ferric thiocyanate method to the determination of incipient rancidity in fats and oils. Oil Soap 1943, 20, 240–243.
- Zhao, T.; Huang, L.; Luo, D.; Xie, Y.; Zhang, Y.; Zhang, Y.; Jiao, W.; Su, G.; Zhao, M. Fabrication and characterization of anchovy protein hydrolysates-polyphenol conjugates with stabilizing effects on fish oil emulsion. Food Chem. 2021, 351, 129324.
- Okada, Y.; Okajima, H.; Konishi, H.; Terauchi, M.; Ishii, K.; Liu, I.M.; Watanabe, H. Antioxidant effect of naturally occurring furan fatty acids on oxidation of linoleic acid in aqueous dispersion. J. Am. Oil Chem. Soc. 1990, 67, 858–862.
- Chen, Y.; Huang, F.; Xie, B.; Sun, Z.; McClements, D.J.; Deng, Q. Fabrication and characterization of whey protein isolates- lotus seedpod proanthocyanin conjugate: Its potential application in oxidizable emulsions. Food Chem. 2021, 346, 128680.
- Jiang, J.; Zhang, Z.; Zhao, J.; Liu, Y. The effect of non-covalent interaction of chlorogenic acid with whey protein and casein on physicochemical and radical-scavenging activity of in vitro protein digests. Food Chem. 2018, 268, 334–341.
- Qie, X.; Chen, Y.; Quan, W.; Wang, Z.; Zeng, M.; Qin, F.; Chen, J.; He, Z. Analysis of beta-lactoglobulin-epigallocatechin gallate interactions: The antioxidant capacity and effects of polyphenols under different heating conditions in polyphenolic-protein interactions. Food Funct. 2020, 11, 3867–3878.
- Dong, H.; Yin, X.; Wusigale; Cheng, H.; Choijilsuren, N.; Chen, X.; Liang, L. Antioxidant activity and stability of alpha-tocopherol, resveratrol and epigallocatechin-3-gallate in mixture and complexation with bovine serum albumin. Int. J. Food Sci. Technol. 2021, 56, 1788–1800.
- Cheng, H.; Fang, Z.; Wusigale; Bakry, A.M.; Chen, Y.; Liang, L. Complexation of trans- and cis-resveratrol with bovine serum albumin, beta-lactoglobulin or alpha-lactalbumin. Food Hydrocoll. 2018, 81, 242–252.
More