Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Hongmei Wang.
The liver serves as a vital organ with a primary metabolic function. In addition, it possesses the ability to synthesize and decompose proteins, regulate overall blood volume, eliminate toxins, and regulate immunity, all of which are crucial for maintaining normal physiological activities in the human body.
- TRP channels
- liver diseases
- liver injury
- liver fibrosis
- hepatocellular carcinoma
1. Transient RPeceptor Potential Channels in Liver Injury
Numerous acute and chronic liver injuries elicit liver inflammation by means of Kupffer cells (KC) activation and recruitment of monocytes and neutrophils, which is facilitated by chemokines. Studies indicate that the hepatic concentrations of chemokines (Ccl2 and Cxcl2) and proinflammatory cytokines (TNF-α, il-1α, il-1β, and il-6) are markedly reduced in TRPV1 knockout mice and that neutrophil infiltration is attenuated [49][1]. Furthermore, there is speculation regarding the potential of the transient receptor potential (TRP) channels to augment the inflammatory response subsequent to hepatocyte injury by regulating the biological activities of KCs, monocytes, and neutrophils, which includes cytokine and chemokine production, chemokine response, adhesion, and migration from the bloodstream to the damaged liver [44][2]. Acetaminophen (APAP) overdose represents a prevalent etiology of acute liver failure in developed nations, frequently necessitating liver transplantation for patient survival. Current studies have shown that TRP channel family members play an important role in APAP-mediated acute liver injury.
Cytochrome P450, located in the ER, catalyzes the conversion of APAP to N-acetyl-para benzoquinone (pBQ) imine (NAPQI). In instances where the accumulation of NAPQI surpasses the capacity for rapid neutralization via glutathione (GSH) coupling, a cascade reaction of liver cell necrosis is triggered through the interaction between NAPQI and multiple downstream proteins [128][3]. On the one hand, NAPQI has the ability to directly stimulate the TRPV4 channel and bind to free sulfhydryl groups located in cysteine residues of the TRPC1 and TRPV1 channels, thereby enhancing their oxidative modification and ultimately resulting in channel opening. On the other hand, NAPQI protein adducts are capable of regulating the function of the respiratory chain, generating peroxides (such as H2O2), which induce mitochondrial oxidative stress, and stimulating the activation of TRPM2 and TRPM7 channels by ROS production [45][4]. Research has indicated that TRPM2 can be transported to the PM of hepatocytes through lysosomal exocytosis in response to oxidative stress induced by H2O2 or APAP. The primary activator of TRPM2 is intracellular ADP ribose (ADPR), which binds to the TRPM2 NUDT9-h motif located at the C-terminus, leading to the opening of the channel pore [89,91,129][5][6][7]. TRPM2 is activated by H2O2 and other oxidants by augmenting the production of ADPR. Simultaneously, the production of intracellular ADPR responds to DNA damage caused by ROS by activating poly ADP ribose (pADPR) polymerase (PARP) [71][8]. The activation of multiple TRP channels expressed by hepatocytes leads to an increase in intracellular Ca2+ concentration, which further aggravates the mitochondrial oxidative stress reaction and finally leads to the activation of RIP3 and translocation of Drp1 and Bax to mitochondria. Bax-induced outer membrane permeability and Drp1-induced mitochondrial fission mediate the mitochondrial membrane permeability transition (MPT). Apoptosis-inducing factor (AIF) and endonuclease are released from the mitochondria and transferred to the nucleus, where they initiate nuclear DNA fragmentation [128][3] (Figure 1). In addition, the harmful effects of TRPM2 activation may be caused by not only Ca2+ entry but also TRPM2-mediated inflow of Na+ and outflow of K+. The accumulation of intracellular Na+ and the loss of K+ lead to the loss of PM potential and the activation of Na+-K+-ATPase, leading to further depletion of cell ATP levels and promoting cell necrosis [130][9].
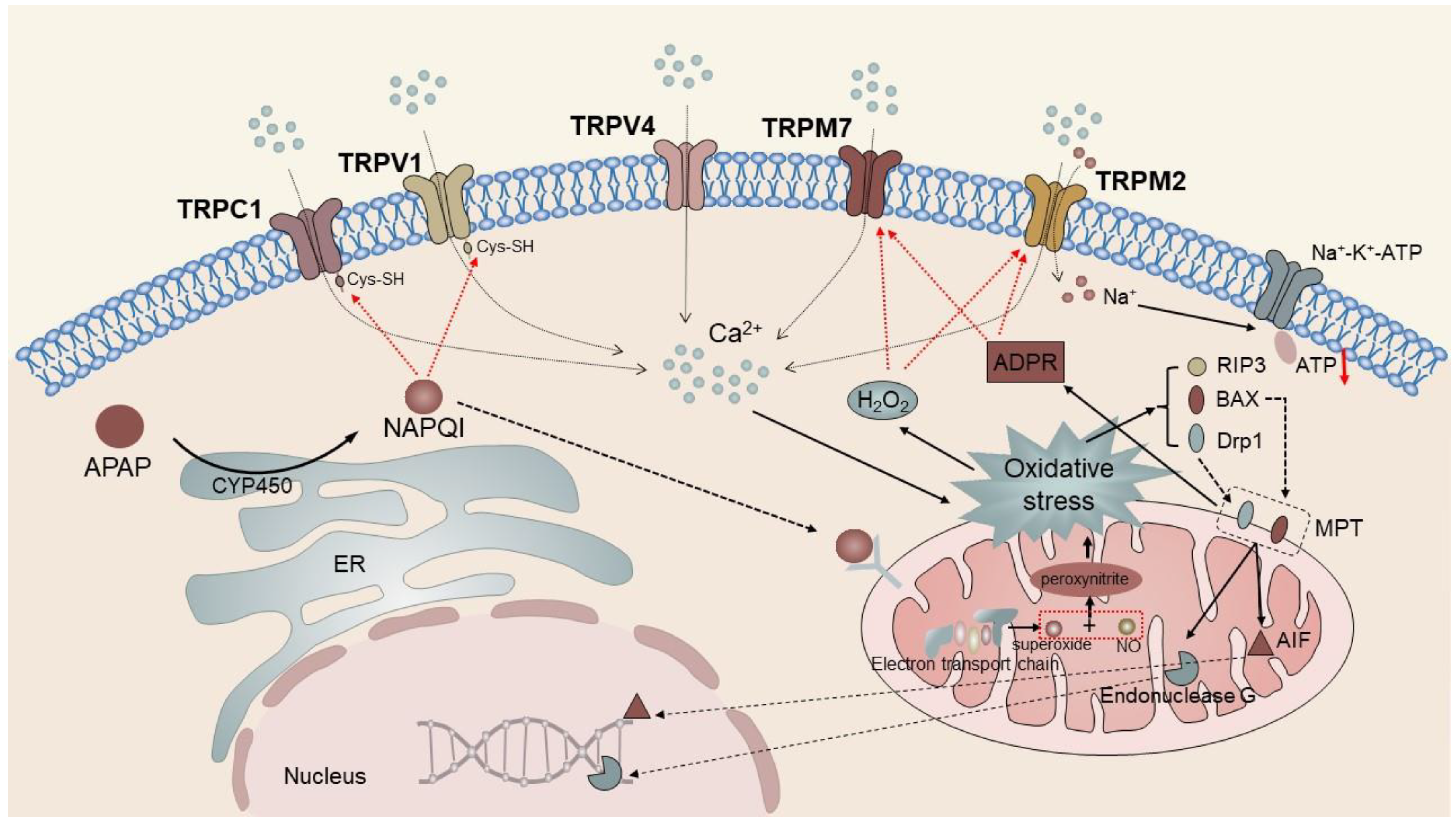
Figure 1. TRPC1, TRPV1, TRPV4, TRPM2, and TPM7 mediate APAP-induced liver injury. The intracellular metabolite of APAP, NAPQI, directly activates TRPC1 and TRPV1 to trigger Ca2+ influx and induce mitochondrial oxidative stress. It eventually leads to the activation of RIP3 and translocation of Drp1 and Bax to the mitochondria, triggering mitochondrial membrane permeability transition. In addition, NAPQI protein adducts modulate respiratory chain function similarly inducing mitochondrial oxidative stress responses. TRPM2 and TRPM7 channels activated by ROS and ADPR further aggravate intracellular cation concentrations, forming a vicious cycle and ultimately causing apoptosis.
N-acetylcysteine (NAC) is the only effective drug therapy for patients with APAP-induced liver injury, and it is considered to be able to supplement and replace the GSH required to bind NAPQI. The GSH inducer DMF attenuates the decrease in GSH content, increase in ROS levels, Ca2+ overload, and cell death-related events induced by APAP or H2O2. Thus, it may be used as a treatment method to replace NAC [45][4]. In addition, because APAP-mediated acute liver injury depends on Ca2+ influx mediated by multiple members of the TRP channel superfamily, it is suggested that targeted inhibition of these TRP channels may be a new therapeutic strategy for acute liver injury.
2. Transient RPeceptor Potential Channels in Liver Fibrosis
Chronic and persistent liver injury resulting from viral infection, excessive alcohol consumption, metabolic etiology, and autoimmune liver disease typically leads to liver fibrosis, a condition marked by the activation of HSCs and excessive accumulation of ECM [131,132][10][11]. During the initial inflammatory stage, fibrosis serves as a beneficial mechanism for wound repair. This process is facilitated by the coordinated activation of coagulation, immune response, and fibroblast activation, which promote the regeneration of damaged tissue or the formation of fibrous scar tissue. However, prolonged inflammation can impede the synthesis, deposition, and degradation of ECM components, leading to fibrosis inhibition. Tissue injury and fibrogenic mediators, such as TGF-β and platelet-derived growth factor (PDGF), mediate the differentiation of stationary HSCs into proliferating myofibroblast-like cells. The activation of HSCs is accompanied by cellular changes, including the loss of vitamin A droplets, de novo expression of α-SMA, and overproduction and deposition of ECM components, such as type I collagen [133][12]. The TRP channels play an important role in the activation of HSCs and the development of liver fibrosis (Table 2).
TRPM7 shows significant permeability toward both Mg2+ and Ca2+ and is a crucial component in maintaining cellular Mg2+ homeostasis. Its expression in HSCs is associated with cell survival. Studies have demonstrated that the inhibition of TRPM7 channels through the use of Gd3+ and 2-aminoethoxydiphenyl borate (2-APB) results in increased apoptosis and reduced expression of α-SMA and Col1α1 in HSC-T6 cells [76,104][13][14]. The downregulation of TRPM7 mediates the inhibition of HSC-T6 cell proliferation and is associated with decreased expression of cyclin D1, cyclin-dependent kinase 4, and proliferating cell nuclear antigen [75][15]. HSCs are responsible for ECM secretion, deposition, and accumulation, and are a crucial contributor to liver fibrosis. Consequently, blocking TRPM7 to impede HSC activation and proliferation and induce apoptosis may represent a promising approach to prevent or reverse liver fibrosis.
Liu et al. used CCl4 and BDL to develop a mouse model of liver fibrosis. Their investigation revealed a notable increase in TRPM8 expression within liver fibrosis tissue. Furthermore, they observed a significant reduction in liver injury and fibrosis in TRPM8−/− mice when compared with the control mice [46][16]. Subsequent investigation has revealed that TRPM8 knockdown results in reduced expression of S100A9, a factor associated with inflammation, and HNF4α, a regulator of gene expression specific to the liver. S100A9 is a constituent of the S100 protein family, which can be discharged from cells undergoing inflammation or damage and binds to toll-like receptor 4 or advanced glycation end product receptor, thereby stimulating an inflammatory reaction. Further, HNF4α is a central transcriptional regulator of hepatocyte gene expression, differentiation, and function maintenance and has been proven to play a central regulatory role in alleviating liver fibrosis [134][17]. The inhibition of TRPM8 has the potential to mitigate liver fibrosis by downregulating the expression of the pro-inflammatory factor S100A9 and promoting the expression of HNF4α. Moreover, the inhibition of TRPM8 has been observed to decrease the number of F4/80-positive cells and suppress the activation of HSCs and cholangiocytes by reducing gene expression levels of IL-6, IL-1β, TNFα, and MCP-1 in a mouse liver fibrosis model. Cholangiocytes, which proliferate in response to endogenous and exogenous stimuli, actively participate in intrahepatic inflammation and repair processes, thereby promoting the progression of liver fibrosis [46,135,136][16][18][19] (Figure 2).
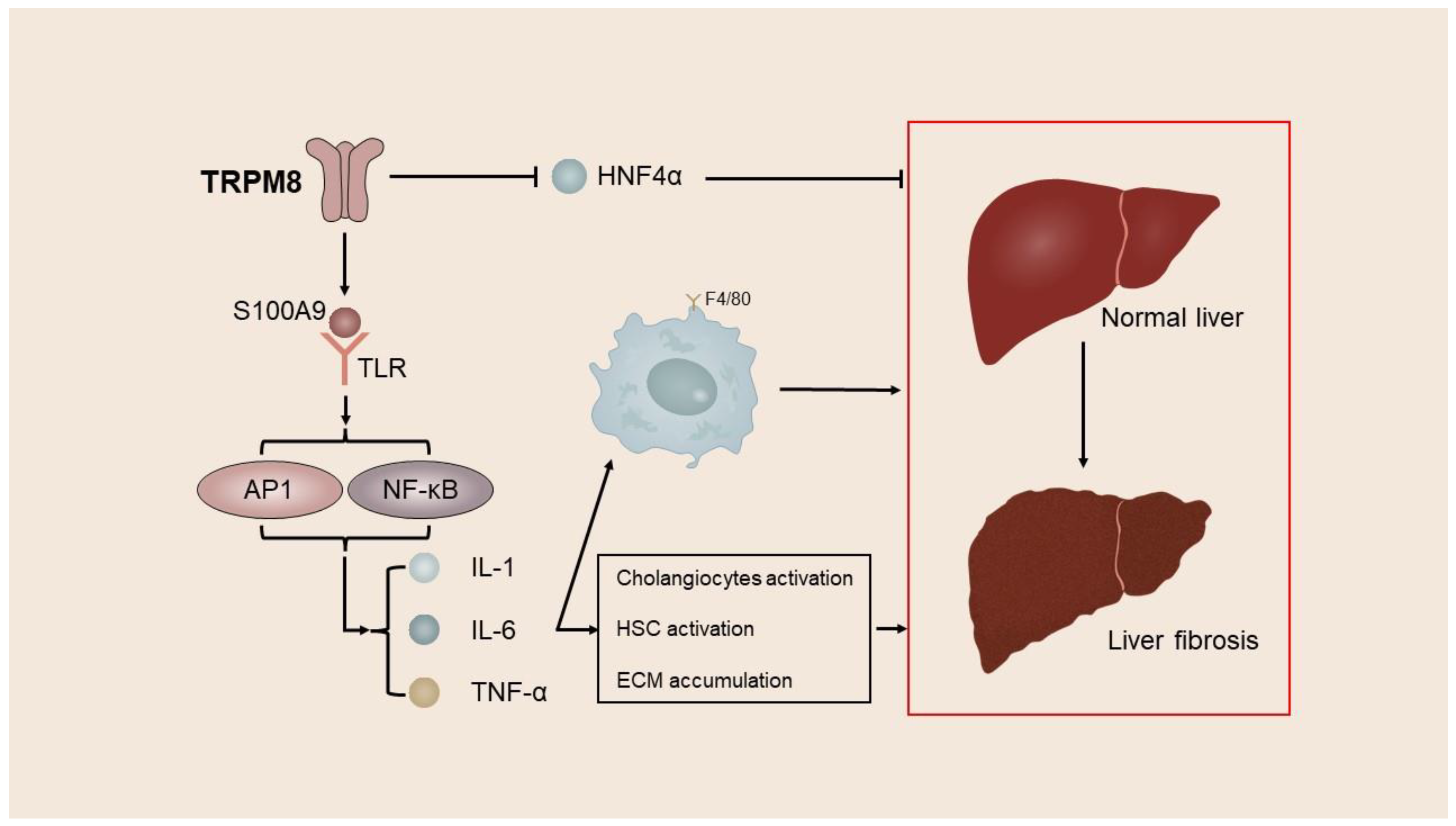
Figure 2. TRPM8 mediates the process of liver fibrosis. Activation of TRPM8 upregulates the inflammation-related factor S100A9 and downregulates the liver-specific gene expression regulator HNF4α while increasing the number of F4/80-positive cells, which in turn promotes the activation of HSCs and cholangiocytes and thereby mediates liver fibrosis.
In a previous in vitro experiment, knockout of TRPV3 with siRNA hindered DNA synthesis and HSC proliferation and increased cell apoptosis [57][20]. Further, activation of TRPV3 has been reported to upregulate the inflammation-related gene Olr1, resulting in increased expression of the protein LOX-1. LOX-1 facilitates the uptake of oxidized low-density lipoprotein (ox-LDL) into endothelial cells and reduces ox-LDL content in the blood. In vivo investigations have demonstrated a positive correlation between LOX-1 expression and inflammatory factor levels [56,137][21][22]. Following TRPV3 activation, a conspicuous infiltration of macrophages has been observed in the area of fibrotic lesions. Macrophage polarization can be induced by various injury factors, and the release of inflammatory factors and chemokines by activated macrophages can exacerbate liver inflammation and fibrosis [138][23]. Consequently, a reduction in TRPV3 expression or functional level may ameliorate the inflammatory response and mitigate the proliferation of fibrotic tissue.
In vivo, the activation of the TRPV4 channel has been observed to exacerbate liver fibrosis, whereas the inhibition of the TRPV4 channel has been shown to alleviate liver fibrosis. Notably, the TRPV4 channel has been found to be significantly upregulated in the tissues of patients with liver fibrosis and CCl4-treated rats. In addition, under the influence of TGF-β1, TRPV4 expression in HSC-T6 cells has been found to increase, potentially because of direct regulation by miR-203 [107][24]. TRPV4 knockdown using siRNA has been demonstrated to strongly inhibit the proliferation of activated HSC-T6 cells, as downregulation of TRPV4 appears to inhibit the autophagy of TGF-β1-treated HSC-T6 cells [59][25]. Autophagy is a crucial mechanism for cellular proliferation and apoptosis and also serves as a catalyst for HSC activation. Suppression of autophagy can impede HSC proliferation and facilitate HSC apoptosis. Upon stimulation with TGF-β1, cultured HSC-T6 cells have previously exhibited significant activation of the AKT signaling pathway. Studies indicate that the AKT signaling pathway is intricately linked to autophagy, and inhibition of the AKT signaling pathway activation can promote HSC apoptosis [139][26]. Thus, TRPV4 may inhibit HSC apoptosis by regulating the activation of the autophagy-dependent AKT signaling pathway, and targeting TRPV4 may become an effective treatment strategy to prevent the progression of liver fibrosis.
The HSC cell line lx-2 cells exhibit a significant upregulation of TRPC6, which is dependent on the activation of NICD under hypoxic conditions. Hypoxia, being an environmental stressor, triggers the activation of oxygen-sensitive HSCs and facilitates the onset of liver fibrosis. The activation of the fiber formation process under hypoxic conditions is mediated by the hypoxia-inducible factor (HIF), which has been identified as a key mediator. Further, the upregulation of hypoxia-inducible factor 1α (HIF1α) has been reported to result in an increase in the nuclear localization of NICD, which subsequently induces the expression of TRPC6. The influx of Ca2+ through TRPC6 channels directly activates the Ca2+-sensitive protein phosphatase calcineurin, which in turn triggers the synthesis of ECM proteins via its downstream transcriptional effector, activated T cell nuclear factor (NFAT) [69][27]. Previous research has demonstrated that Smad3 plays a significant role in the fibrotic response of HSCs, and it is believed that TRPC6 promotes the expression of α-SMA and collagen through the activation of SMAD2/3 [140][28] (Figure 3).

Figure 3. TRPC6 mediates the process of liver fibrosis. TRPC6 is significantly upregulated under hypoxic conditions in a manner dependent on NICD activation. TRPC6 promotes α-SMA and collagen expression through the activation of calcineurin and SMAD2/3, leading to liver fibrosis.
To conclude, the upregulation of TRPM7, TRPM8, TRPV3, TRPV4, and TRPC6 is significantly associated with the onset and progression of liver fibrosis. Targeting these TRP channels in clinical settings may offer a potential strategy for the prevention or reversal of liver fibrosis, thereby safeguarding the liver against the deleterious effects of advanced cirrhosis and HCC.
3. Transient RPeceptor Potential Channels in Liver Cancer
The predominant forms of liver cancer are HCC and intrahepatic cholangiocarcinoma, with HCC comprising approximately 90% of all liver cancer cases. Other types of liver cancer include mixed hepatocellular cholangiocarcinoma, fibrolamellar HCC, pediatric hepatoblastoma, and metastatic liver cancer [141][29]. HCC is the sixth most prevalent cancer type and the third leading cause of cancer-related death. Because of the liver’s robust compensatory capacity, HCC is frequently detected in intermediate and advanced stages, resulting in suboptimal surgical resection opportunities and diminished therapeutic outcomes. Furthermore, the persistent high recurrence rate of HCC poses a formidable challenge, rendering it to be a significant global health concern [142,143][30][31]. Hence, there is a pressing need to identify novel biological markers and therapeutic targets for the timely detection and improved prognosis of HCC. Given the prevalence of HCC in liver cancer and the focus of this investigation, this researticle ch solely presents an overview of the expression and function of TRP channels in HCC.
Recent research suggests that cancer may be classified as a channel disease, as tumor cell survival, death, and movement are regulated through ion channels and transporters. Of particular significance is the role of Ca2+ as a crucial messenger in the regulation of cell proliferation, apoptosis, transcription, migration, and angiogenesis. Studies have demonstrated that the disruption of intracellular Ca2+ homeostasis creates a unique microenvironment within the liver, facilitating the activation of various signaling pathways, including Wnt/bcatenin, TP53/cell cycle, telomere maintenance, chromatin regulatory factors, and others. These pathways drive mutagenesis, leading to the accelerated proliferation of hepatocytes [144,145,146][32][33][34]. TRP channels play a significant role in regulating various cellular physiological and pathophysiological processes in cancer, primarily by modulating the expression levels of functional TRP proteins to dynamically regulate intracellular ion concentrations, rather than through TRP gene mutations. Notably, multiple TRP channels have been identified in HCC at both the gene and protein levels. Specifically, both Huh7 and HepG2 human hepatoma cell lines express mRNAs for TRPC1, TRPC6, TRPV1, TRPV2, TRPV4, TRPM4, TRPM6, TRPM7, and TRPM8, whereas Huh7 alone expresses TRPV3 and TRPM5 [85][35].
3.1. Transient RPeceptor Potential Canonical
TRPC1 is co-located with lipid raft proteins, such as caveolin-1, and is considered to be an important component of SOCE. SOCE can maintain the Ca2+ levels in the ER. When SOCE is inhibited, it lacks storage and supplementation, and cell growth and proliferation are inhibited [147][36]. Cancer cell migration has been reported to be induced by TGF-β through the stimulation of intracellular Ca2+ release and Ca2+ entry via TRPC1 and Na+/Ca2+ exchangers. Further, TRPC1 silencing resulted in the inhibition of Huh7 cell proliferation, while both Ca2+ release and SOCE in the ER were upregulated, suggesting that TRPC1 exerts a regulatory effect on SOCE. The overexpression of TRPC1 in HCC is linked to unfavorable prognoses of afflicted patients. Bioinformatics analysis suggests that TRPC1 impedes retinol metabolism and other vital metabolic processes by amplifying shared signaling pathways that facilitate tumor proliferation and the expression of genes such as ABI2, MAPRE1, YEATS2, MTA3, and TMEM237 [148][37]. Following TRPC1 silencing, there was a significant decrease in the expression of cell cycle-regulating genes CDK11A/11B and URGCP, accompanied by a notable increase in the expression of survival-promoting genes ERBB3 and FGFR4 [149,150][38][39]. Similarly, TRPC1 inhibition through shRNA or SKF 96365 in D54MG glioma cells resulted in a reduction in cell proliferation. In vivo experiments have further demonstrated that the inhibition of TRPC1 expression led to a reduction in the size of lateral abdominal tumors [151][40].
TRPC5 exhibits high expression levels in paracancerous tissues, and its activation is potentially mediated by regulation of the Akt/IκB/NF-κB signaling pathway, which inhibits macrophage differentiation and causes an increase in M1 macrophage infiltration. The release of proinflammatory cytokines and chemokines (such as TNF-α, IL-12, and iNOS) by M1 macrophages results in the inhibition of cell proliferation, reduction in tumor cell invasiveness, and suppression of malignant phenotypes, which ultimately leads to the promotion of favorable prognoses for patients with HCC [66,67,68][41][42][43].
The present study arch reveals that the expression of TRPC6 mRNA and protein is significantly upregulated in liver cancer tissues compared with normal liver tissues. Furthermore, in vitro experiments using HepG2 and Huh7 cells demonstrate that TGF-β plays a crucial role in the development of HCC by promoting the formation of the TRPC6-NCX1 complex, which leads to an elevation in intracellular Ca2+ levels and subsequently enhances the migratory and invasive properties of HCC cells [47][44]. Overexpression of TRPC6 in HCC cells results in the continuous accumulation of intracellular free calcium, which in turn stimulates epithelial–mesenchymal transition (EMT), Hif1-α signal transduction, and DNA damage repair. This process enables HCC cells to acquire multi-drug resistance (MDR), which may be mediated by the calcium-dependent TRPC6/calcium/STAT3 pathway. The use of siRNA or SKF 96365 to inhibit TRPC6 can alleviate MDR induced by various stimuli. Further, targeted inhibition of TRPC6 in combination with adriamycin in vivo demonstrates a synergistic anti-tumor effect [152][45]. Furthermore, significant inhibition of HCC cell proliferation can be achieved by blocking TRPC6. This suggests that TRPC6 targeting can potentially serve as a novel anti-tumor approach by alleviating MDR and suppressing HCC proliferation.
3.2. TRPVransient Receptor Potential vanilloid
Research has demonstrated that the activation of TRPV1 by capsaicin is responsible for the elevation in intracellular Ca2+ levels, production of ROS, and activation of the STAT3 pathway in HepG2 cells. This presents a promising avenue for targeting and eliminating HCC cells as well as regulating their migration [153][46]. MMP1 upregulation in HCC patients is noteworthy because of its association with poor prognosis. However, the activation of TRPV1 can increase MMP1 expression, which contradicts previous studies linking TRPV1 overexpression to longer disease-free survival in liver cancer patients [48,58,154,155][47][48][49][50]. Currently, no study has established a direct correlation between TRPV1 expression and MMP1 upregulation in HCC cells, necessitating further research to elucidate the underlying mechanism by which TRPV1 regulates HCC cells.
During the progression from hepatitis to cirrhosis to liver cancer, there is a gradual increase in both TRPV2 mRNA and protein levels. In contrast, poorly differentiated tumors exhibit reduced expression of TRPV2 mRNA and protein compared with well-differentiated HCC tumors. In addition, a correlation exists between TRPV2 expression and portal vein invasion, thereby suggesting that TRPV2 serves as a potent prognostic marker for patients with HCC [52][51]. ROS facilitates the upregulation of TRPV2 mRNA and protein levels in HepG2 and Huh7 cells, resulting in the suppression of survival-promoting factors, including Akt and Nrf2, and the stimulation of death-promoting factors, namely, p38 and JNK1, during the initial phase of apoptosis [53][52]. Furthermore, the expression of TRPV2 has been found to be associated with the desiccation of HCC cells. Notably, the downregulation of TRPV2 has been observed to significantly enhance the colony-forming capacity of HepG2 cells and the expression of stem cell markers CD133 and CD44 in hepatoma cell lines. Conversely, the overexpression of TRPV2 has been shown to diminish the formation of globules and colonies [54][53]. Hagit et al. presented a novel approach to enhance the clinical application of doxorubicin, a chemotherapeutic drug, in the liver cancer cell line BNL1 ME. They revealed that the co-administration of cannabinol (CBD) or 2-APB with doxorubicin resulted in significant augmentation of doxorubicin accumulation in BNL1 ME cells [55][54]. The limited penetration of doxorubicin through the cell membrane due to its weak alkaline chemical nature led to a concentration gradient, with the concentration of doxorubicin outside the cell being approximately three times higher than that inside the cell [156][55]. The clinical utility of doxorubicin is constrained by its irreversible cardiac toxicity. The BNL1 ME cell line, derived from mice with HCC, exhibits the expression of the macroporous cation channel receptor TRPV2. The agonists of TRPV2, namely, CBD and 2-APB, facilitate the activation of TRPV2, thereby facilitating the entry and accumulation of doxorubicin within BNL1 ME cells. In addition, CBD has been demonstrated to inhibit P-gp ATPase, thereby reducing the efflux of doxorubicin from cells [157][56]. Furthermore, the absence of noteworthy TRPV2 protein expression in cardiomyocytes or hepatocytes implies that localized hepatic administration of this drug combination will exclusively affect HCC cells without any off-target consequences. However, the non-specific TRPV2 agonists, 2-APB and CBD, may trigger other ion channels, thereby disturbing the ion homeostasis in normal cells [158][57] (Figure 4).
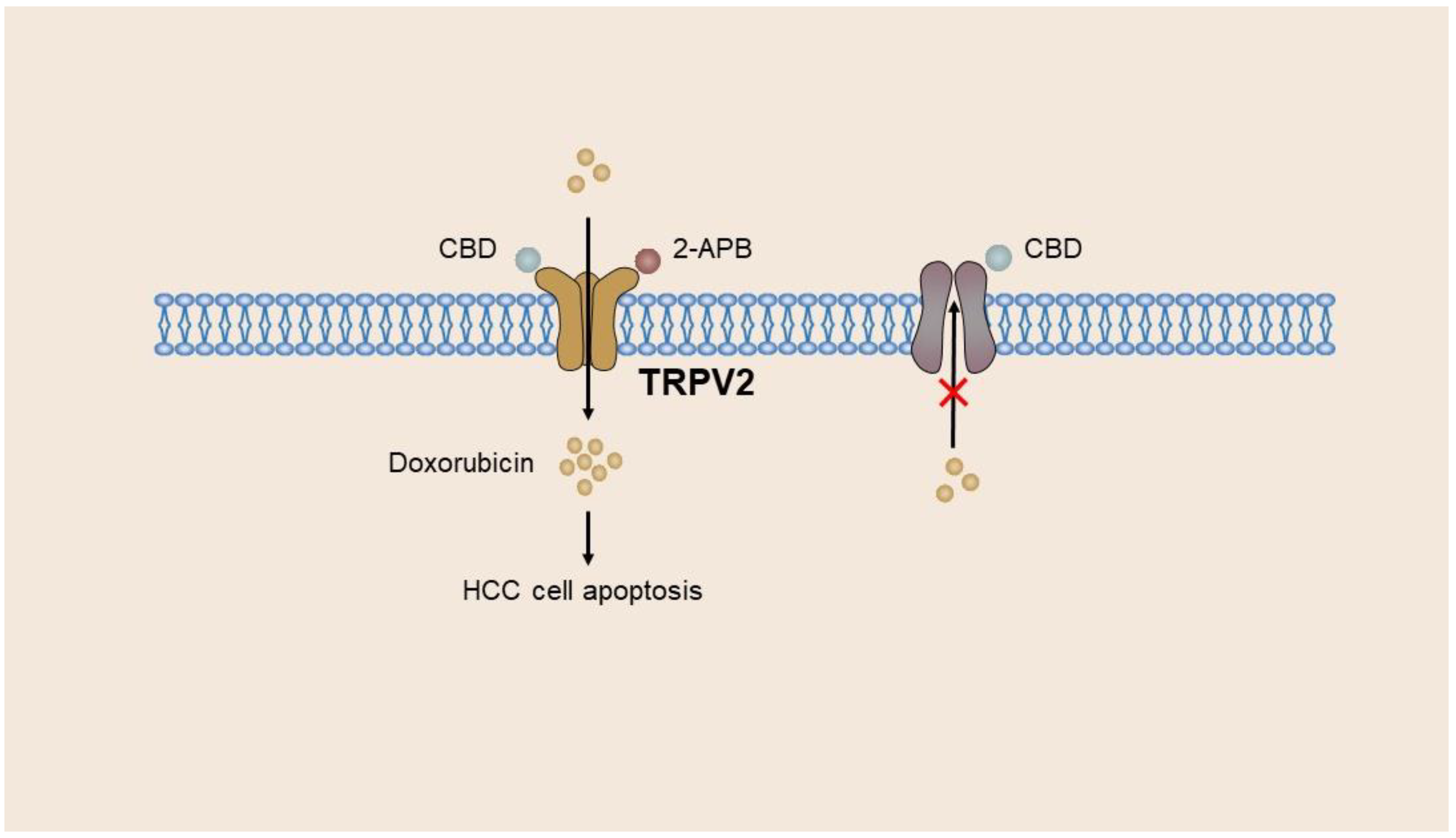
Figure 4. Adriamycin enters BNL1 ME cells of the hepatoma cell line through TRPV2. CBD and 2-APB act as agonists of TRPV2 to activate and open the TRPV2 channel, promoting the entry and massive accumulation of doxorubicin in BNL1 ME cells. Meanwhile, CBD inhibits P-gp ATPase, thereby reducing doxorubicin removal from cells.
In vitro, the TRPV4 channel of HCC cells was inhibited by the specific antagonist HC067047, resulting in the inhibition of cell proliferation and promotion of cell apoptosis. This effect was achieved with the weakening of EMT and inactivation of p-ERK. The pro-apoptotic effect of TRPV4 was found to be blocked by influencing the transcription and translation of apoptosis-related genes Bax and Bcl2, ultimately leading to the activation of caspase 3 [159][58]. Similarly, Lee et al. reported that TRPV4 inhibitors can effectively inhibit the migration and invasion of 4T07 human breast cancer cells with high expression of TRPV4 [160][59].
Venom peptide (Tv1) treatment in HCC cell line 1’s MEP cells resulted in a significant reduction in COX-2 and prostaglandin E2 (PGE2) levels, which was found to be associated with TRPV6/TRPC6-mediated calcium-dependent apoptosis [40][60]. The dephosphorylation of calcineurin activates the Ca2+-dependent transcription factor NFAT, which regulates COX-2 expression in various cancer cells [161,162][61][62]. Overexpression of COX-2 results in elevated levels of PGE2, which subsequently binds to the EP receptor. The activation of the EP receptor in turn is responsible for facilitating angiogenesis, inhibiting apoptosis, stimulating cell growth, and enhancing the potential for invasion and metastasis of tumor tissue [163][63]. TV1 has the potential to impede the influx of Ca2+ into neoplastic cells via TRPV6 by interacting with extracellular recruitment sites comprising E518, E519, and N548 residues in the TRPV6 tetramer and via selective filters located in the D542 side chain of the pore region. This mechanism may effectively hinder the aforementioned process of tumor initiation and advancement [40][60] (Figure 5).
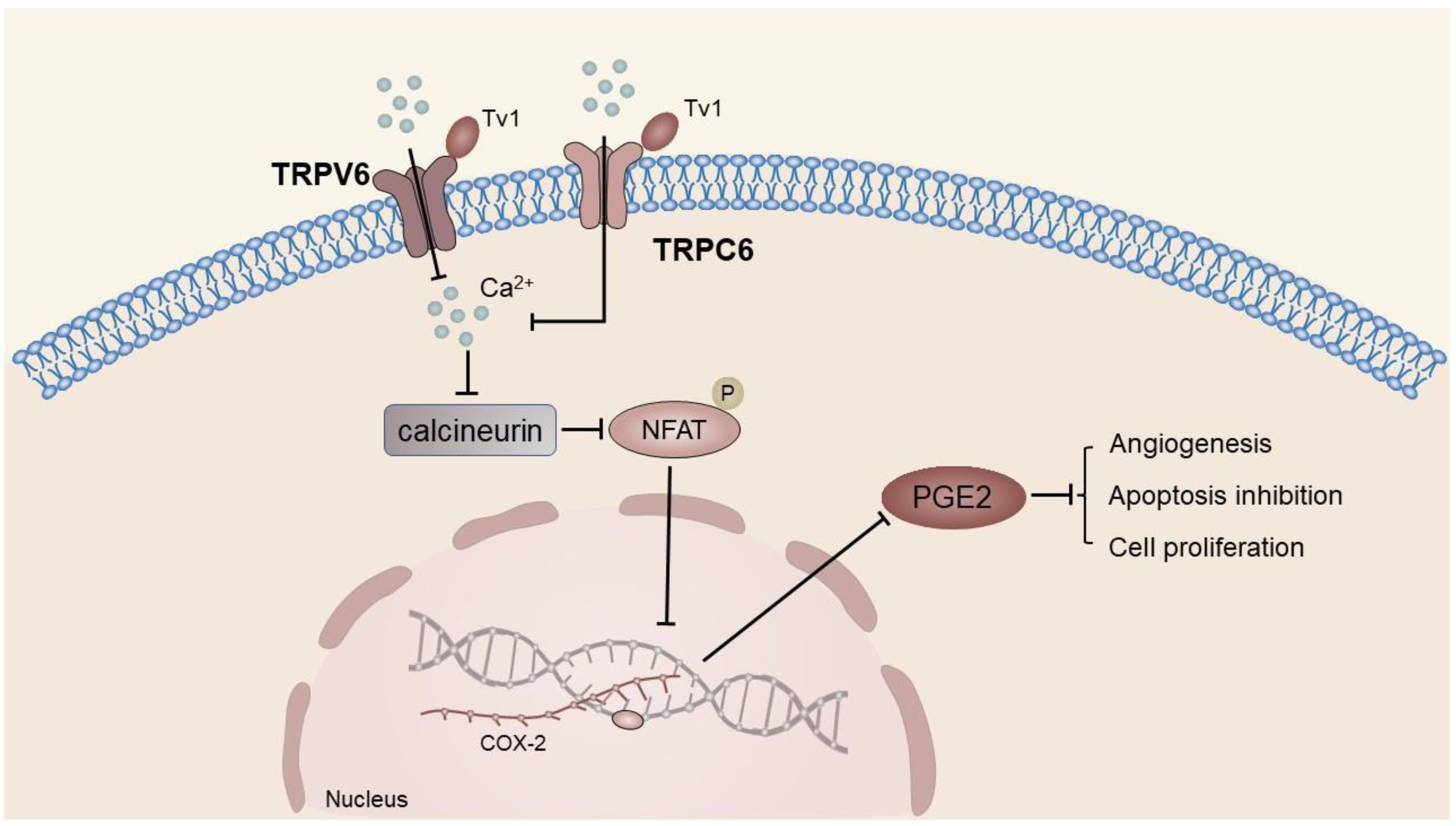
Figure 5. COX-2 and PGE2 levels were significantly decreased in venom peptide (Tv1)-treated 1MEA cells. Tv1 prevented Ca2+ influx after binding to TRPV6 and TRPC6, and the transcription factor NFAT could not be dephosphorylated by calcineurin, thus downregulating COX-2 expression.
The combination of drugs targeting the COX-2/PGE2 axis and traditional anti-tumor drugs presents a promising approach for liver cancer treatment. Specifically, the combination of selective COX-2 inhibitor meloxicam and T7 polypeptide has a potent anti-tumor effect on liver cancer in mice [164][64]. Nevertheless, the multifaceted physiological and pathological functions of PGE2 necessitate caution in the use of COX-2 inhibitors, as long-term intake may result in adverse effects on the kidney and cardiovascular system [165,166][65][66]. Hence, there is an urgent need to devise novel targets. Despite the incomplete elucidation of the TRPV6 mechanism that hinders apoptosis of hepatoma cells, it presents fresh perspectives for investigators. The potential for TRP channel-targeting drugs to supplement selective COX-2 inhibitors in cancer therapy necessitates additional exploration.
3.3. Transient RPeceptor Potential Melastatin
Functional TRPM7-like channels have been identified in the proliferating and polarized rat hepatoma cell line WIF-B, and their regulation is dependent on the concentration of cytoplasmic Ca2+ through CaMKII-dependent processes. These channels may contribute to the survival of WIF-B cells and may also be involved in the migration of hepatoma cells. In particular, TRPM7-mediated Ca2+ influx has been shown to enhance the activity of calpain, which is known to play a role in the migration of HepG2 cells [167][67] (Table 1).
Table 1.
The role of TRP channels in liver-related diseases.
| Liver Diseases | TRP Channels | Function | Ref. |
|---|---|---|---|
| Hepatic injury | TRPV1, TRPV4, TRPC1, TRPM2, TRPM7 | Enhance the inflammatory response after hepatocyte injury | [44,45,89,91][2][4][5][6] |
| Hepatic fibrosis | TRPM7, TRPM8, TRPV3, TRPV4, TRPC6 | Promote the activation and proliferation of HSCs and aggravate liver fibrosis | [46,1357,59,][1476,][16104,]140,168][[20][25][28][68] |
| HCC | TRPC1, TRPC6, TRPV6, TRPV4, TRPM7 | Promote the progression of HCC | [40,47,148,153,159,167,169][37][44][46][58][60][67][69] |
| TRPC5, TRPV1, TRPV2 | Reduce the aggressiveness of tumor cells | [52,,68][4155,][4266,][43]67[51][54] |
Abbreviations: HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells.
4. Drugs for TRPransient receptor potential Channels
Numerous drugs function by targeting TRP channels during the treatment of diseases. As TRP is extensively expressed in the sensory neurons, brain, and skin, earlier studied drugs were formulated to alleviate chronic pain. For instance, the TRPV1 agonist capsaicin (8-methyl-N-vanillyl-6-nonenamide), possessing analgesic and anesthetic properties, is frequently used to mitigate post-therapeutic neuralgia, diabetic neuralgia, rheumatoid arthritis pain, and osteoarthritis pain. Nevertheless, a drawback of TRPV1 agonists is their potential to elicit a robust initial pain response, thereby constraining their clinical application. Consequently, non-stimulatory TRPV1 agonists, including N-(3-methoxy-4-hydroxybenzyl) oleamide (NE19550) and MDR-652, have been investigated by researchers to circumvent these untoward effects. However, these compounds are currently undergoing animal experimentation and require further clinical validation [170][70]. An ultrapotent capsaicin analog, resiniferatoxin, has demonstrated efficacy in alleviating pain in canines afflicted with osteosarcoma, as well as in human subjects diagnosed with metastatic cervical cancer, and is presently undergoing clinical trials. Researchers aim to achieve sustained pain relief for individuals suffering from severe osteoarthritis and chronic cancer [17][71]. In the context of TRPV1 receptor-targeted drugs, a therapeutic dose of a TRPV1 agonist functions as an antagonist of the TRPV1 receptor. In particular, a low dose of capsaicin stimulates the release of inflammatory mediators via TRPV1, whereas a therapeutic dose inhibits such a release. Consequently, the initial generation of TRPV1 antagonists was associated with febrile reactions and burns [18][72]. However, the second-generation TRPV1 antagonist JNJ-39439335 (mavatrep) has demonstrated significant efficacy in reducing pain during exercise in patients with knee arthritis in clinical trials [171][73]. Eucalyptol (1,8-cineol), another TRPV1 antagonist, has demonstrated pain relief in mice with gouty arthritis, whereas the TRPV1 antagonist SB-366791 (N-(3-methoxyphenyl)-4-chlorocinn amide) exhibited pain relief in rats with tooth pain. In addition to TRPV1-targeting drugs, TRPA1 antagonists, such as A-967079, have shown the potential to treat neuropathic pain in animal experiments. Furthermore, GRC17536 (Glenmark Pharmaceuticals), a TRPA1 antagonist that is currently undergoing clinical trials, has been used to alleviate diabetic neuropathic pain. Other investigations have indicated that small-molecule TRPM8 antagonists may alleviate cold-induced pain [17][71].
TRPV1 antagonists, such as SB-705498 (N-(2-bromophenyl)-N’-[((R)-1-(5-trifluoromethyl-2-pyridyl) pyrrolidin-3-yl)]urea), have the potential to mitigate capsaicin-induced cough in addition to pain management. The reversal of acetylcholine-induced airway hyperreactivity in mice with Ovalbumin-induced allergic asthma with the TRPA1 antagonist HC-030031 indicates the therapeutic potential of TRPA1 in the inhibition of asthma. Further, the oral TRPA1 antagonist GDC-0334 has demonstrated efficacy in suppressing cough, airway hyperresponsiveness, and edema in clinical trials. In contrast, menthol, an over-the-counter cough suppressant, acts as a TRPM8 agonist and may effectively inhibit citric acid-induced cough by activating TRPM8 [17][71]. The TRPV1 agonists, capsaicin and resiniferatoxin, have been found to have potential applications beyond pain management. In particular, they may be used to desensitize individuals with a neurogenic bladder, resulting in increased bladder capacity and reduced instances of incontinence [172][74]. In addition, topical resiniferatoxin desensitization may be used for the prevention of premature ejaculation in males [173][75]. The present study investigated the efficacy of TRPV1 antagonists, namely, GRC-6211, and TRPM8 antagonists, namely, RQ-00434739 and KRP-2529, in mitigating hyperactivity in a chronically inflamed bladder. In addition, the TRPV4 agonist, GSK1016790A, was examined for its potential to improve underactive bladders, and TRPV4 antagonists were evaluated for their ability to ameliorate overactive bladders [174][76]. TRP drugs have demonstrated potential for application in dermatology. Notably, the TRPV1 antagonist PAC-14028, currently in phase III clinical trials, has been found to improve the skin barrier function and alleviate pruritus in patients with atopic dermatitis. However, topical administration of this drug may increase the risk of infection in patients. Nonetheless, the TRPM8 agonist menthoxypropanediol cream has been shown to alleviate pruritus in humans, while the topical administration of the TRPA1 antagonist HC-030031 has been found to alleviate UV-induced burn injury in mice. Furthermore, TRPV4 inhibition has also been found to alleviate skin pruritus. TRP-targeted drugs exhibit potential in treating ocular and central nervous system diseases. In animal experiments, the TRPV4 antagonist HC-067047 was effective in treating ocular matrix opacities following alkali burn [175][77]. Similarly, the TRPM2 antagonist JNJ-28583113 has shown promising results in improving cognitive dysfunction in mice after cerebral ischemia [17][71].
TRP-targeting drugs have demonstrated the ability to enhance glucose metabolism. Specifically, in murine models of type 2 diabetes, the small molecule antagonist N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl) tetrahydropyrazine-1(2H)-carbox-amide (BCTC) targeting TRPV1 has been found to improve insulin secretion in response to glucose stimulation. In addition, clinical trials have shown that the TRPV1 antagonist XEN-D0501, when used in conjunction with metformin, exhibits favorable glucose-lowering effects [176][78]. The efficacy of numerous drugs in cancer treatment is linked to TRP channels. For instance, Vacquinol-1, an immunosuppressant, triggers cell death in glioblastoma cells via its TRPM7 ATP-inducible inhibitory effect. Another example is gemcitabine, a cytotoxic drug that exhibits heightened cytotoxicity when combined with anti-TRPM7 siRNA, which may be triggered through the p16 (CDKN2A) and WRN mRNA pathway [151][40].
Animal studies have validated the association between TRP channels and organ fibrosis. In particular, the administration of TRPC6 antagonist BI 749327 has been shown to effectively impede the progression of kidney and myocardial fibrosis. This is achieved by suppressing the activation of the nuclear factor of activated T cells (NFAT), thereby hindering the expression of pro-hypertrophic genes [177][79]. Despite the extensive application of TRP-targeting drugs in animal testing and clinical trials, there remains a paucity of drugs used for liver diseases. Current research efforts are concentrated on treating liver fibrosis. In mouse experiments, the TRPM8 antagonist M8-B hydrochloride exhibited potential in inhibiting the activation of cholangiocytes by downregulating the expression of inflammatory factor S100A9 and upregulating the expression of HNF4α, which ultimately led to the amelioration of hepatobiliary inflammation and liver fibrosis [46][16]. Furthermore, investigations conducted on a CCl4-induced liver fibrosis mouse model demonstrated that the TRPV4 agonist GSK1016790A exacerbated liver fibrosis, whereas the TRPV4 antagonist HC-067047 effectively ameliorated liver collagen fiber deposition and the degree of liver lobular disorder [178][80]. Similarly, drofenine, a TRPV3 selective agonist, exacerbated liver fibrosis in a CCl4-induced mouse liver fibrosis model, while forsythoside B, a TRPV3 inhibitor, significantly mitigated liver fibrosis, although the underlying mechanism warrants further investigation [57][20]. TRP channels have potential as a drug target for the treatment of liver fibrosis and drug-induced hepatotoxic injury. The inhibition of TRPV4, for instance, has been shown to mitigate the hepatotoxic side effects resulting from excessive APAP in mice with acute liver failure [130][9].
References
- Liu, H.; Beier, J.I.; Arteel, G.E.; Ramsden, C.E.; Feldstein, A.E.; McClain, C.J.; Kirpich, I.A. Transient receptor potential vanilloid 1 gene deficiency ameliorates hepatic injury in a mouse model of chronic binge alcohol-induced alcoholic liver disease. Am. J. Pathol. 2015, 185, 43–54.
- Wang, W.; Liu, P.; Zhang, Y.; Yan, L.; Zhu, M.X.; Wang, J.; Yu, Y. Expression and functions of transient receptor potential channels in liver diseases. Acta Pharm. Sin. B 2023, 13, 445–459.
- Ramachandran, A.; Jaeschke, H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J. Clin. Transl. Res. 2017, 3, 157–169.
- Badr, H.; Kozai, D.; Sakaguchi, R.; Numata, T.; Mori, Y. Different Contribution of Redox-Sensitive Transient Receptor Potential Channels to Acetaminophen-Induced Death of Human Hepatoma Cell Line. Front. Pharmacol. 2016, 7, 26903865.
- Toth, B.; Csanady, L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J. Biol. Chem. 2010, 285, 30091–30102.
- Iordanov, I.; Mihalyi, C.; Toth, B.; Csanady, L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife 2016, 5, e17600.
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599.
- Kheradpezhouh, E.; Zhou, F.H.; Barritt, G.J.; Rychkov, G.Y. Oxidative stress promotes redistribution of TRPM2 channels to the plasma membrane in hepatocytes. Biochem. Biophys. Res. Commun. 2018, 503, 1891–1896.
- Echtermeyer, F.; Eberhardt, M.; Risser, L.; Herzog, C.; Gueler, F.; Khalil, M.; Engel, M.; Vondran, F.; Leffler, A. Acetaminophen-induced liver injury is mediated by the ion channel TRPV4. FASEB J. 2019, 33, 10257–10268.
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol.-Mech. Dis. 2011, 6, 425–456.
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669.
- Moreira, R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007, 131, 1728–1734.
- Liu, H.; Li, J.; Huang, Y.; Huang, C. Inhibition of transient receptor potential melastain 7 channel increases HSCs apoptosis induced by TRAIL. Life Sci. 2012, 90, 612–618.
- Zhu, Y.; Men, R.; Wen, M.; Hu, X.; Liu, X.; Yang, L. Blockage of TRPM7 channel induces hepatic stellate cell death through endoplasmic reticulum stress-mediated apoptosis. Life Sci. 2014, 94, 37–44.
- Fang, L.; Zhan, S.; Huang, C.; Cheng, X.; Lv, X.; Si, H.; Li, J. TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol. Appl. Pharmacol. 2013, 272, 713–725.
- Liu, Q.; Lei, X.; Cao, Z.; Zhang, J.; Yan, L.; Fu, J.; Tong, Q.; Qin, W.; Shao, Y.; Liu, C.; et al. TRPM8 deficiency attenuates liver fibrosis through S100A9-HNF4α signaling. Cell Biosci. 2022, 12, 58.
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296.
- Park, S.M. The crucial role of cholangiocytes in cholangiopathies. Gut Liver 2012, 6, 295–304.
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577.
- Yan, L.; Zhang, X.; Fu, J.; Liu, Q.; Lei, X.; Cao, Z.; Zhang, J.; Shao, Y.; Tong, Q.; Qin, W.; et al. Inhibition of the transient receptor potential vanilloid 3 channel attenuates carbon tetrachloride-induced hepatic fibrosis. Biochem. Biophys. Res. Commun. 2021, 558, 86–93.
- Chen, M.; Nagase, M.; Fujita, T.; Narumiya, S.; Masaki, T.; Sawamura, T. Diabetes enhances lectin-like oxidized LDL receptor-1 (LOX-1) expression in the vascular endothelium: Possible role of LOX-1 ligand and AGE. Biochem. Biophys. Res. Commun. 2001, 287, 962–968.
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77.
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Letteron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2019, 68, 522–532.
- Song, Y.; Zhan, L.; Yu, M.; Huang, C.; Meng, X.; Ma, T.; Zhang, L.; Li, J. TRPV4 channel inhibits TGF-beta1-induced proliferation of hepatic stellate cells. PLoS ONE 2014, 9, e101179.
- Zhan, L.; Yang, Y.; Ma, T.; Huang, C.; Meng, X.; Zhang, L.; Li, J. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol. Cell. Biochem. 2015, 402, 9–22.
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470.
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126.
- Iyer, S.C.; Kannan, A.; Gopal, A.; Devaraj, N.; Halagowder, D. Receptor channel TRPC6 orchestrate the activation of human hepatic stellate cell under hypoxia condition. Exp. Cell Res. 2015, 336, 66–75.
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428.
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6.
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314.
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/beta-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926.
- Nakagawa, H.; Fujita, M.; Fujimoto, A. Genome sequencing analysis of liver cancer for precision medicine. Semin. Cancer Biol. 2019, 55, 120–127.
- Montagner, A.; Le Cam, L.; Guillou, H. beta-catenin oncogenic activation rewires fatty acid catabolism to fuel hepatocellular carcinoma. Gut 2019, 68, 183–185.
- El, B.C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077.
- Gill, D.L.; Waldron, R.T.; Rys-Sikora, K.E.; Ufret-Vincenty, C.A.; Graber, M.N.; Favre, C.J.; Alfonso, A. Calcium pools, calcium entry, and cell growth. Biosci. Rep. 1996, 16, 139–157.
- Qi, H.; Wu, F.; Wang, H. Function of TRPC1 in modulating hepatocellular carcinoma progression. Med. Oncol. 2023, 40, 97.
- Selli, C.; Erac, Y.; Kosova, B.; Erdal, E.S.; Tosun, M. Silencing of TRPC1 regulates store-operated calcium entry and proliferation in Huh7 hepatocellular carcinoma cells. Biomed. Pharmacother. 2015, 71, 194–200.
- Selli, C.; Pearce, D.A.; Sims, A.H.; Tosun, M. Differential expression of store-operated calcium- and proliferation-related genes in hepatocellular carcinoma cells following TRPC1 ion channel silencing. Mol. Cell. Biochem. 2016, 420, 129–140.
- Zhong, T.; Zhang, W.; Guo, H.; Pan, X.; Chen, X.; He, Q.; Yang, B.; Ding, L. The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies. Acta Pharm. Sin. B 2022, 12, 1761–1780.
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell. Biochem. 2018, 119, 9419–9432.
- Ye, Y.; Xu, Y.; Lai, Y.; He, W.; Li, Y.; Wang, R.; Luo, X.; Chen, R.; Chen, T. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J. Cell. Biochem. 2018, 119, 2951–2963.
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 803037.
- Xu, J.; Yang, Y.; Xie, R.; Liu, J.; Nie, X.; An, J.; Wen, G.; Liu, X.; Jin, H.; Tuo, B. The NCX1/TRPC6 Complex Mediates TGFbeta-Driven Migration and Invasion of Human Hepatocellular Carcinoma Cells. Cancer Res. 2018, 78, 2564–2576.
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q.; et al. Regulation of Multi-drug Resistance in hepatocellular carcinoma cells is TRPC6/Calcium Dependent. Sci. Rep. 2016, 6, 23269.
- Chen, W.T.; Lin, G.B.; Lin, S.H.; Lu, C.H.; Hsieh, C.H.; Ma, B.L.; Chao, C.Y. Static magnetic field enhances the anticancer efficacy of capsaicin on HepG2 cells via capsaicin receptor TRPV1. PLoS ONE 2018, 13, e191078.
- Waning, J.; Vriens, J.; Owsianik, G.; Stüwe, L.; Mally, S.; Fabian, A.; Frippiat, C.; Nilius, B.; Schwab, A. A novel function of capsaicin-sensitive TRPV1 channels: Involvement in cell migration. Cell Calcium 2007, 42, 17–25.
- Vriens, J.; Janssens, A.; Prenen, J.; Nilius, B.; Wondergem, R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 2004, 36, 19–28.
- Dai, L.; Mugaanyi, J.; Cai, X.; Dong, M.; Lu, C.; Lu, C. Comprehensive bioinformatic analysis of MMP1 in hepatocellular carcinoma and establishment of relevant prognostic model. Sci. Rep. 2022, 12, 13639.
- Li, W.H.; Lee, Y.M.; Kim, J.Y.; Kang, S.; Kim, S.; Kim, K.H.; Park, C.H.; Chung, J.H. Transient receptor potential vanilloid-1 mediates heat-shock-induced matrix metalloproteinase-1 expression in human epidermal keratinocytes. J. Investig. Dermatol. 2007, 127, 2328–2335.
- Liu, G.; Xie, C.; Sun, F.; Xu, X.; Yang, Y.; Zhang, T.; Deng, Y.; Wang, D.; Huang, Z.; Yang, L.; et al. Clinical significance of transient receptor potential vanilloid 2 expression in human hepatocellular carcinoma. Cancer Genet. Cytogenet. 2010, 197, 54–59.
- Ma, W.; Li, C.; Yin, S.; Liu, J.; Gao, C.; Lin, Z.; Huang, R.; Huang, J.; Li, Z. Novel role of TRPV2 in promoting the cytotoxicity of H2O2-mediated oxidative stress in human hepatoma cells. Free Radic. Biol. Med. 2015, 89, 1003–1013.
- Hu, Z.; Cao, X.; Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Cao, Z.; Du, H.; Cheng, X.; et al. Transient receptor potential vanilloid-type 2 targeting on stemness in liver cancer. Biomed. Pharmacother. 2018, 105, 697–706.
- Neumann-Raizel, H.; Shilo, A.; Lev, S.; Mogilevsky, M.; Katz, B.; Shneor, D.; Shaul, Y.D.; Leffler, A.; Gabizon, A.; Karni, R.; et al. 2-APB and CBD-Mediated Targeting of Charged Cytotoxic Compounds Into Tumor Cells Suggests the Involvement of TRPV2 Channels. Front. Pharmacol. 2019, 10, 1198.
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677.
- Zhu, H.J.; Wang, J.S.; Markowitz, J.S.; Donovan, J.L.; Gibson, B.B.; Gefroh, H.A.; Devane, C.L. Characterization of P-glycoprotein inhibition by major cannabinoids from marijuana. J. Pharmacol. Exp. Ther. 2006, 317, 850–857.
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.J. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273.
- Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Liu, Q.; Liu, G.; Cao, Z.; Fu, J.; Du, H.; et al. Pharmacological inhibition of TRPV4 channel suppresses malignant biological behavior of hepatocellular carcinoma via modulation of ERK signaling pathway. Biomed. Pharmacother. 2018, 101, 910–919.
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6, 27903.
- Anand, P.; Filipenko, P.; Huaman, J.; Lyudmer, M.; Hossain, M.; Santamaria, C.; Huang, K.; Ogunwobi, O.O.; Holford, M. Selective Inhibition of Liver Cancer Cells Using Venom Peptide. Mar. Drugs 2019, 17, 587.
- Qu, L.; Liu, B. Cyclooxygeanse-2 promotes metastasis in osteosarcoma. Cancer Cell Int. 2015, 15, 69.
- Hai, L.; Kawarabayashi, Y.; Imai, Y.; Honda, A.; Inoue, R. Counteracting effect of TRPC1-associated Ca2+ influx on TNF-alpha-induced COX-2-dependent prostaglandin E2 production in human colonic myofibroblasts. Am. J. Physiol.-Gastroint. Liver Physiol. 2011, 301, G356–G367.
- Nzeako, U.C.; Guicciardi, M.E.; Yoon, J.H.; Bronk, S.F.; Gores, G.J. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 2002, 35, 552–559.
- Yang, J.; Zhong, J.; Zhou, M.; Zhou, Y.; Xiu, P.; Liu, F.; Wang, F.; Li, Z.; Tang, Y.; Chen, Y.; et al. Targeting of the COX-2/PGE2 axis enhances the antitumor activity of T7 peptide in vitro and in vivo. Drug Deliv. 2021, 28, 844–855.
- Sharma, J.N.; Jawad, N.M. Adverse effects of COX-2 inhibitors. Sci. World J. 2005, 5, 629–645.
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA-J. Am. Med. Assoc. 2001, 286, 954–959.
- Chen, Y.; Yu, Y.; Sun, S.; Wang, Z.; Liu, P.; Liu, S.; Jiang, J. Bradykinin promotes migration and invasion of hepatocellular carcinoma cells through TRPM7 and MMP2. Exp. Cell Res. 2016, 349, 68–76.
- Sun, B.; Zhang, X.; Cheng, X.; Zhang, Y.; Chen, L.; Shi, L.; Liu, Z.; Qian, H.; Wu, M.; Yin, Z. Intratumoral hepatic stellate cells as a poor prognostic marker and a new treatment target for hepatocellular carcinoma. PLoS ONE 2013, 8, e80212.
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010, 70, 418–427.
- Ann, J.; Kim, H.S.; Thorat, S.A.; Kim, H.; Ha, H.J.; Choi, K.; Kim, Y.H.; Kim, M.; Hwang, S.W.; Pearce, L.V.; et al. Discovery of Nonpungent Transient Receptor Potential Vanilloid 1 (TRPV1) Agonist as Strong Topical Analgesic. J. Med. Chem. 2020, 63, 418–424.
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59.
- Moran, M.M.; Szallasi, A. Targeting nociceptive transient receptor potential channels to treat chronic pain: Current state of the field. Br. J. Pharmacol. 2018, 175, 2185–2203.
- Manitpisitkul, P.; Flores, C.M.; Moyer, J.A.; Romano, G.; Shalayda, K.; Tatikola, K.; Hutchison, J.S.; Mayorga, A.J. A multiple-dose double-blind randomized study to evaluate the safety, pharmacokinetics, pharmacodynamics and analgesic efficacy of the TRPV1 antagonist JNJ-39439335 (mavatrep). Scand. J. Pain 2018, 18, 151–164.
- Phe, V.; Schneider, M.P.; Peyronnet, B.; Abo, Y.N.; Mordasini, L.; Chartier-Kastler, E.; Bachmann, L.M.; Kessler, T.M. Intravesical vanilloids for treating neurogenic lower urinary tract dysfunction in patients with multiple sclerosis: A systematic review and meta-analysis. A report from the Neuro-Urology Promotion Committee of the International Continence Society (ICS). Neurourol. Urodyn. 2018, 37, 67–82.
- Shi, B.; Li, X.; Chen, J.; Su, B.; Li, X.; Yang, S.; Guan, Z.; Wang, R. Resiniferatoxin for treatment of lifelong premature ejaculation: A preliminary study. Int. J. Urol. 2014, 21, 923–926.
- Deruyver, Y.; Weyne, E.; Dewulf, K.; Rietjens, R.; Pinto, S.; Van Ranst, N.; Franken, J.; Vanneste, M.; Albersen, M.; Gevaert, T.; et al. Intravesical Activation of the Cation Channel TRPV4 Improves Bladder Function in a Rat Model for Detrusor Underactivity. Eur. Urol. 2018, 74, 336–345.
- Okada, Y.; Shirai, K.; Miyajima, M.; Reinach, P.S.; Yamanaka, O.; Sumioka, T.; Kokado, M.; Tomoyose, K.; Saika, S. Loss of TRPV4 Function Suppresses Inflammatory Fibrosis Induced by Alkali-Burning Mouse Corneas. PLoS ONE 2016, 11, e167200.
- Gram, D.X.; Fribo, J.; Nagy, I.; Gotfredsen, C.; Charrua, A.; Hansen, J.B.; Hansen, A.J.; Szallasi, A. TRPV1 Antagonists as Novel Anti-Diabetic Agents: Regulation of Oral Glucose Tolerance and Insulin Secretion Through Reduction of Low-Grade Inflammation? Med. Sci 2019, 7, 82.
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del, C.D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161.
- Fu, J.; Du, H.; Zhang, X.; Xu, X. Pharmacological Inhibition of Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Alleviates Carbon Tetrachloride-Induced Liver Fibrosis in Mice. J. Nippon Med. Sch. 2019, 86, 258–262.
More