Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
The roles of gut microbiota are highly regarded in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). The intestinal bacteria regulate the metabolism of bile acids depending on bile salt hydrolase (BSH), 7-dehydroxylation, hydroxysteroid dehydrogenase (HSDH), or amide conjugation reaction, thus exerting effects on NAFLD development through bile acid receptors such as farnesoid X receptor (FXR), Takeda G-protein-coupled bile acid protein 5 (TGR5), and vitamin D receptor (VDR), which modulate nutrient metabolism and insulin sensitivity via interacting with downstream molecules. Reversely, the composition of gut microbiota is also affected by the level of bile acids in turn.
- bile acid
- bile acid receptor
- gut microbiota
- NAFLD
- NAFLD treatment
1. The Mechanism of How Gut Microbiota Influences the Progression of Nonalcoholic Fatty Liver Disease through Bile Acids
1.1. Farnesoid X Receptor
1.1.1. Gut Microbiota Have an Impact on Nonalcoholic Fatty Liver Disease through Bile Acid–Farnesoid X Receptor Pathway
Farnesoid X receptor (FXR) is highly expressed in the liver and ileum. In the liver, bile acid-activated FXR inhibits the expression of cholesterol 7-alpha- hydroxylase (CYP7A1) via the induction of small heterodimer partner (SHP; NR0B2). CYP7A1 is generally considered the rate-limiting enzyme that initiates bile acid synthesis. The activation of FXR in the distal ileum induces the expression of Fgf15 (FGF19 in humans), which binds to the FGF receptor 4 (FGFR4)/β-klotho heterodimer complex when it reaches the liver by portal blood to inhibit CYP7A1 expression [1]. FGF15/19 could also inhibit bile acid generation by increasing the stability of SHP [2]. Bile acid affinities for FXR are as follows: CDCA > LCA = DCA > CA [3]. Phe-chol and Tyr-chol are also strong human FXR agonists. It is proven that Phe-chol is an agonist twice as strong as CDCA. These bile acids could increase the expression of the FXR effector genes Fgf15 in the intestine and Shp in both the intestine and liver [4].
Research about cholesterol gallstone disease finds that the bacterium Desulfovibrionales is associated with enhanced cecal secondary bile acid production and is enriched in patients with gallstone disease. H2S is the product of Desulfovibrionales that could induce hepatic FXR and inhibit CYP7A1 expression, which results in the accumulation of cholesterol in the liver [5]. Activating hepatic FXR could ameliorate NASH according to research about the application of disulfiram in the treatment of NASH. Disulfiram inhibits the growth of Clostridium and reduces Clostridium-mediated 7α-dehydroxylation activity to suppress secondary bile acid biosynthesis, which in turn activates hepatic FXR signaling to ameliorate NASH [6]. Hepatocyte MyD88 ablation exhibits an increase in Ruminococcus and Oscillospira and a decrease in Sutterella and Allobaculum, resulting in the decrease in the main FXR agonist CA and the increase in FXR antagonist T-βMCA, thereby contributing to glucose intolerance, inflammation, and hepatic insulin resistance in mice [7].
Interestingly, activation of the FXR in the intestine seems to have the opposite effect on nonalcoholic fatty liver disease (NAFLD). Intestine-specific Fxr disruption ameliorates hepatic triglyceride accumulation in mice under a high-fat diet [8]. Theabrownin reduces BSH-enriched bacteria and increases the levels of ileal conjugated bile acids to inhibit the intestinal FXR-FGF15 signaling pathway, resulting in increased hepatic production and fecal excretion of bile acids, which in turn reduces hepatic cholesterol and decreases lipogenesis [9]. Type 2 diabetes (T2D) patients treated with metformin revealed a decrease in Bacteroides fragilis and an increase in FXR antagonist glycoursodeoxycholic acid (GUDCA), which supports the beneficial effect of intestinal FXR antagonist to metabolic dysfunction [10]. The Western diet induces an increase in the abundance of Firmicutes and a relative reduction in the abundance of Bacteroides, which mediates significantly increased DCA and LCA. DCA-mediated FXR activation in the myeloid cells in the intestine can lead to the production and activation of type I IFN resulting in the dysfunction of intestinal Paneth cells and intensifying gut inflammation in mice [11]. Another study has proven that hepatic thyroid hormone signaling modulates glucose homeostasis through repressing intestinal FXR signaling [12].
However, there are opposing conclusions concerning the effect of intestinal FXR agonists.
Lactobacillus rhamnosus GG-treated mice showed a reduction in taurine-β-muricholic acid (T-βMCA), an FXR antagonist, and the amelioration of liver inflammation and fibrosis, while these changes are reversed by intestine-specific FXR inhibitors. It proves that increasing the intestinal FXR signaling pathway leads to the suppression of bile acid de novo synthesis and prevents excessive bile acid-induced liver injury and fibrosis in mice [13]. And both liver and intestine FXR agonists could reduce bacterial translocation via the portal-venous route to the liver in cirrhosis [14]. Application of the intestine-restricted FXR agonist fexaramine (FEX) markedly increases taurolithocholic acid (TLCA) and improves metabolism indicators in db/db mice. FEX increases the transformation of LCA from CDCA by amplifying the amount of Acetatifactor and Bacteroides, which have high BSH and 7α- and 7β-dehydroxylase activity. LCA then activates TGR5 signaling to stimulate GLP-1 secretion from L cells [15], resulting in promoting adipose tissue browning, improving hepatic insulin signaling and glucose metabolism [16]. Another research study found increased levels of total and taurine-conjugated bile acid pool sizes and intestinal FXR signaling after Roux-en-Y gastric bypass (RYGB) surgery, which is linked to increased TGR5 expression and stimulates adaptive thermogenesis in rats [17]. As there is a frequent substance exchange between the gut and brain, it is intriguing to find that inhibition of FXR in the dorsal vagal complex enhances insulin action in HFD-fed rats [18].
In conclusion, gut microbiota transforms bile acid composition to regulate FXR distributed in the different locations and influences the metabolism status in NAFLD as shown in Figure 1.
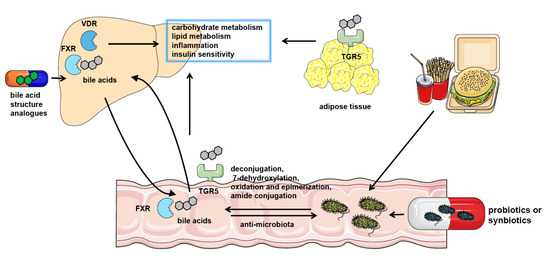
Figure 1. The crosstalk between gut microbiota and bile acids in NAFLD. NAFLD patients have significantly altered gut microbiota composition and bile acid pool. Gut microbiota catalyzes bile acid metabolism via deconjugation, 7-dehydroxylation, oxidation, and epimerization or amide conjugation. The bile acids could also regulate the constituents of the gut microbiome by the detergent actions. Bile acids play a role in regulating nutrient metabolism in NAFLD by binding with bile acid receptors such as FXR, VDR, and TGR5 in the liver, intestine, and adipocytes or other tissues. And pharmacological treatment using probiotics or synbiotics or applying analogs of bile acids are effective approaches to the therapy of NAFLD.
1.1.2. The Molecular Mechanism of How Farnesoid X Receptor Influences Nonalcoholic Fatty Liver Disease
The expression of FXR is suppressed by iron [19]. FXR transcription activation could also be regulated by glucose via FXR own protein O-GlcNAcylation [20]. The histone deacetylase Sirtuin (SIRT) overexpression causes deacetylation of FXR with low FXR protein expression. And it could also regulate FXR target gene transcription by influencing histone methylation [21].
In NAFLD, FXR activation induces the expression of transporters that provide outflow routes for bile acid efflux to avoid toxic bile acid overload, such as bile acid export pump (BSEP) [22] and organic solute transporter-alpha and -beta (OSTα/β). The inhibition of ASBT by FXR in the enterocytes also prevents the uptake of bile acids [23]. It also upregulates the expression of ATP-binding cassette sub-family G member 5/8 (ABCG5/8) that is in charge of cholesterol efflux [24]. Accordingly, intestinal FXR activation renders the bile acid pool more hydrophylic and less efficient in emulsifying lipids, resulting in cholesterol fecal elimination via ABCG5/8 [25].
FXR activation results in a reduction in sterol regulatory binding protein-1c (SREBP-1c) expression by the FXR-SHP axis and represses hepatic de novo lipogenesis [26]. The increased expression of SHP after FXR activation suppresses hepatocyte nuclear factor 4α (HNF4α) activities. HNF-4 promotes the expression of microsomal triglyceride transfer protein (MTP), which is involved in transferring triglycerides, cholesterol esters, and phospholipids to newly synthesized apolipoprotein (apo) B [27]. FXR could also decrease apolipoprotein CIII (apo CIII) promoter activity [28] and induce the gene expression of apoC-II [29]. It is known that apo CIII inhibits triglyceride lipolysis whereas apo CII is responsible for triglyceride hydrolysis in chylomicrons and very low-density lipoprotein (VLDL). FXR also regulates glucose metabolism via the upregulation of SHP and represses gluconeogenic gene glucose-6-phosphatase (G6Pase) expression [30]. On the other hand, the activation of FXR also represses the expression of the liver-type pyruvate kinase gene (L-PK) through interacting with carbohydrate-responsive element binding protein (ChREBP) and HNF4α to result in the release of these transcription factors from the promoter of L-PK and repressing hepatic glycolysis [31]. In addition, FXR activation protects NAFLD by blunting hepatic inflammation. FXR interacts with NLRP3 and caspase 1, thereafter preventing NLRP3 inflammasome assembly and activation in macrophages [32], which is contradicted with the increased inflammation caused by the elevation of type I IFN after FXR activation in the myeloid cells in the intestine [11] (Figure 2). Moreover, FXR activation in hepatic stellate cells (HSCs) is reported to alleviate liver fibrosis via the inhibition of TGF-β/SMAD3 in a SHP-dependent way [33] or triggering the expression of anti-fibrotic genes, like peroxisomal proliferator-activated receptor γ (PPAR γ) [34].
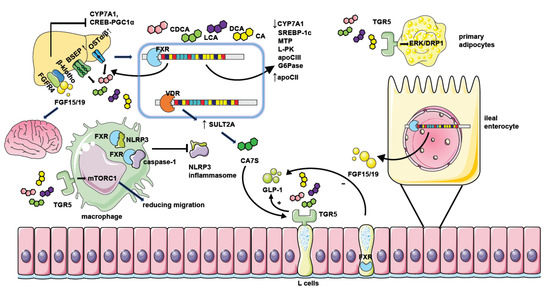
Figure 2. The mechanism of NAFLD regulation by bile acid receptors. Hepatic FXR activation suppresses bile acid synthesis by inhibiting CYP7A1 expression. Intestinal FXR activation could also inhibit CYP7A1 by inducing the generation of FGF15/19 and modulating gene transcription of CYP7A1 through binding with FGFR4/β-klotho receptors in the liver. Hepatic FXR activation promotes bile acid efflux by increasing the expression of bile acid transporters such as BSEP and OSTα/β thus ameliorating the toxic effect of excessive bile acids. FXR activation when binding with bile acids leads to decreased expression of genes involved in lipid or glucose metabolism including SREBP-1c, MTP, L-PK, apoCIII, and G6Pase. It could also repress the CREB-PGC1α pathway by inducing FGF15/19. Together, these factors result in reduced hepatic triglyceride accumulation and improved insulin sensitivity. Moreover, FGF15/19 could also facilitate weight loss by combining with nerve cell receptors. FXR activation in macrophages inhibits the assembling of the NLRP3 inflammasome and ameliorates hepatic inflammation in NAFLD. The activation of TGR5 in L cells causes the secretion of GLP-1 to potentiate insulin secretion, and the activation of TGR5 in primary adipocytes enhances ERK-DRP1 to increase energy expenditure. However, the activation of FXR in L cells inhibits GLP-1 secretion. TGR5 activation in macrophages also suppresses inflammation response by manipulating mTORC1. Hepatic VDR activation increases the generation of CA7S by promoting SULT2A, which stimulates TGR5 in L cells to promote GLP-1 secretion.
FGF15/19, the effector of FXR activation, could also regulate metabolism directly. It could inhibit hepatic gluconeogenesis by inhibiting the cAMP regulatory element-binding protein (CREB)-peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) pathway [35]. FGF19 treatment prevents the accumulation of lipid droplets in Fxr-null mice [36] and inhibits lipogenic enzyme expression by elevation in SHP expression [37]. Moreover, FGF15/19 could reach the brain and mediate beneficial weight loss [38][39]. But knockdown of the obligate coreceptor mediating FGF15/19 signaling named β-klotho shows resistance to diet-induced obesity in mice [40].
1.2. Takeda G-protein-Coupled Bile Acid Protein 5
1.2.1. Gut Microbiota Have an Impact on Nonalcoholic Fatty Liver Disease through Bile Acid-G-Protein-Coupled Bile Acid Protein 5 Pathway
TGR5 is a cell surface receptor belonging to the GPCR, which is expressed in enteroendocrine L cells, white and brown adipose tissue, skeletal muscle, gallbladder, non-parenchymal liver cells, and the brain [41]. The order of activation of TGR5 by bile acids is LCA > DCA > CDCA > CA [41]. In addition to bile acids, some steroid hormone intermediates, such as pregnandiol, are also the ligands of TGR5 [42].
Oligofructose could reduce body weight gain and improve glucose metabolism in mice fed a Western-style diet. It enriches bacteria belonging to Lachnospiraceae and Eggerthellaceae families, which are strongly correlated with cecal HDCA levels. HDCA is elevated after oligofructose administration and it activates TGR5 to stimulate GLP-1 secretion to relieve metabolism disorder [43]. Research shows that intestinal hypoxia-inducible factor 2α (HIF-2α) ablation in mice leads to lower lactate levels by controlling the expression of intestinal Ldha, which in turn results in less Bacteroides vulgatus and greater Ruminococcus torques abundance. Together, these changes elevate taurine-conjugated cholic acid (TCA) and DCA levels and activate TGR5, contributing to the elevation of white adipose tissue thermogenesis [44]. Bloom of Akkermansia muciniphila and a reduction in Lactobacillus in high fat/high sucrose (HFHS)-fed mice result in an increase in α/β-murocholic acid (MCA), DCA, and UDCA, which activate TGR5 and increase energy expenditure [45]. Akkermansia muciniphila is significantly decreased in lean individuals with T2D than their counterparts, which also has a negative correlation with serum 3β-CDCA levels and a positive correlation with insulin secretion and FGF15/19 concentrations [46].
1.2.2. The Molecular Mechanism of How Takeda G-Protein-Coupled Bile Acid Protein 5 Influences Nonalcoholic Fatty Liver Disease
GLP-1 is secreted by enteroendocrine L cells and subsequently potentiates postprandial insulin secretion by pancreatic β cells. TGR5-mediated GLP-1 secretion is dependent on the cAMP mechanism [15]. TGR5 is found to activate mTORC1 signaling in several studies [47][48] and TGR5-mTORC1 signaling in the ileum is also considered to regulate the secretion of GLP-1 [47]. However, FXR activation in L cells decreases the secretion of GLP-1 by interfering with the glucose-responsive factor ChREBP [49] and inhibiting SCFAs induced GLP-1 secretion by decreasing free fatty acid receptor 2 (FFAR2) expression [50]. TGR5 activation in primary adipocytes is necessary for the respiration increase induced by mitochondrial fission through activation of extracellular signal-regulated kinase (ERK)/dynamin-1-like protein (DRP1) signaling [51]. It is reported that TGR5 activation in macrophages reduces macrophage migration depending on mTORC1 [48], thus ameliorating liver inflammation in NAFLD. TGR5 activation in hepatocytes also negatively regulates the hepatic inflammatory response by suppressing the NF-κB pathway by the mediation of the interaction between IκBα and β-arrestin2 [52].
2. The Therapeutic Methods Targeting Microbiota–Bile Acid Pathway
2.1. The Clinical Application of Microbiota in Nonalcoholic Fatty Liver Disease Treatment
Considering the significant effect of the gut microbiota on NAFLD, fecal microbiota transplantation (FMT) has been tested in patients with metabolic syndrome and obesity. Although the improvement of insulin sensitivity is reported, mild alteration of other metabolic parameters is observed [53]. However, specific probiotic treatments show satisfactory clinical efficacy. The mixture of eight probiotic strains (Streptococcus thermophilus, bifidobacteria [B. breve, B. infantis, B. longum], Lactobacillus acidophilus, L. plantarum, L. paracasei, and L. delbrueckii subsp. bulgaricus) decreases the BMI of obese children with biopsy-proven NAFLD and increases the level of GLP-1 [54]. Lactobacillus rhamnosus strain GG treatment reveals a significant decrease in alanine aminotransferase in children with obesity-related liver disease [55]. Another research study using similar bacteria including Streptococcus thermophilus, Bifidobacterium breve, Bifidobacterium longum, Lactobacillus acidophilus, Lactobacillus bulgaricus, Lactobacillus rhamnosus, and Lactobacilluscasei combined with lifestyle modification reports the amelioration of liver inflammation in patients with NAFLD [56]. The beneficial effect of Lactobacilli, Bifidobacteria, and Streptococcus thermophilus on NAFLD has been validated by various clinical research studies [54][56][57][58][59][60]. Moreover, the application of bacteriophages (phages) targeting specific harmful bacteria in NAFLD is a new potential treatment approach. The success of administrating phages against cytolytic E. faecalis to reduce the severity of ethanol-induced liver disease in the mice provides the first evidence that strategies to target single gut microbes might be developed for treatment of NAFLD [61].2.2. The Clinical Application of Bile Acid or Bile Acid Receptor Agonists in Nonalcoholic Fatty Liver Disease Treatment
Therapies using natural bile acids, such as UDCA, have been proven to be successful in the therapy for PBC (primary biliary cirrhosis). However, it has only limited therapeutic efficacy in NASH in several randomized controlled studies [62][63]. The UDCA-homolog norursodeoxycholic acid (norUDCA) seems to be effective for NAFLD treatment with a significant reduction in serum ALT in the clinical trial [64]. Aramchol is a novel synthetic lipid molecule obtained by conjugating two natural components, CA (bile acid) and arachidic acid (saturated fatty acid), through a stable amide bond. It could significantly inhibit the activity of a gene involved in lipid synthesis named stearoyl coenzyme A desaturase 1 (SCD1). The clinical trial confirmed its positive effect on the reduction in liver fat content in NAFLD patients [65]. The impairment of intestinal bile acid absorption by inhibiting ASBT improves features and insulin sensitivity of NAFLD in HFD-fed mice [66]. In phase 1 studies, the ABST inhibitor volixibat could decrease the level of total and low-density lipoprotein (LDL) cholesterol [67]. However, in the double-blind, phase II study, volixibat had no therapeutic impact on steatosis or liver injury in NASH [68]. Further studies on ASBT inhibitors need to be performed to further assess the efficacy and safety of this approach in humans. FXR-specific activator OCA (6α-ethylCDCA) is proven to lead to weight loss and improve histological changes in clinical trials of NASH [69][70]. Other FXR agonists such as cilofexor, tropifexor, and MET409 could reduce hepatic fat content in NASH patients [71][72][73]. However, OCA therapy is associated with an increase in VLDL and LDL particles and a reduction in HDL [74], which may originate from the ability of FXR activation to blunt bile acid synthesis and LDL clearance via repression of CYP7A1. Moreover, FXR activation upregulates the expression of flavin mono-oxygenase3 (FMO3), which oxidized the microbial metabolite of choline and carnitine to trimethylamineN-oxide (TMAO) to contribute to the development of atherosclerosis [75]. So rigorous measurements of the side effects on the cardiovascular system should be taken when using FXR agonists. Considering the beneficial effect of FGF15/19, the FGF19 analog is created and tested for the treatment of NAFLD. Although aberrant FGF19-FGFR4 signaling has been identified in hepatocellular carcinoma [76], using non-tumorigenic FGF19 analog NGM282 produces rapid and significant reductions in liver fat content in NASH patients in the phase II clinical trial without promoting carcinogenesis [77]. There is limited clinical evidence about the effect of the TGR5 activator on the treatment of NAFLD. TGR5 agonist SB-756050 has been tested in T2D patients, but it shows highly variable pharmacodynamic effects in the subjects. It might be explained by the short duration of exposure (6 days) and the small number of subjects (51 patients) [78]. Notably, TGR5 agonists might be toxic to cardiomyocytes [79] and it is proved by a study in which TGR5 agonists result in reflex tachycardia in dogs [80]. TGR5 agonists also increase gallbladder volume in mice [81]. However, there is also an experiment proving the beneficial effect of TGR5 agonists on the cardiovascular system [82]. Larger-scale clinical trials are needed to evaluate the effect of TGR5 agonists in NAFLD treatment. Until now, other bile acid-stimulating receptors such as PXR, CAR, and VDR have not been subjected to anti-NAFLD agent development [83].References
- Gonzalez, F.J.; Jiang, C.; Patterson, A.D. An Intestinal Microbiota–Farnesoid X Receptor Axis Modulates Metabolic Disease. Gastroenterology 2016, 151, 845–859.
- Miao, J.; Xiao, Z.; Kanamaluru, D.; Min, G.; Yau, P.M.; Veenstra, T.D.; Ellis, E.; Strom, S.; Suino-Powell, K.; Xu, H.E.; et al. Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 2009, 23, 986–996.
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365.
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129.
- Hu, H.; Shao, W.; Liu, Q.; Liu, N.; Wang, Q.; Xu, J.; Zhang, X.; Weng, Z.; Lu, Q.; Jiao, L.; et al. Gut microbiota promotes cholesterol gallstone formation by modulating bile acid composition and biliary cholesterol secretion. Nat. Commun. 2022, 13, 252.
- Lei, Y.; Tang, L.; Chen, Q.; Wu, L.; He, W.; Tu, D.; Wang, S.; Chen, Y.; Liu, S.; Xie, Z.; et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat. Commun. 2022, 13, 6862.
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Ståhlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632.
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402.
- Huang, F.; Zheng, X.; Ma, X.; Jiang, R.; Zhou, W.; Zhou, S.; Zhang, Y.; Lei, S.; Wang, S.; Kuang, J.; et al. Theabrownin from Pu-erh tea attenuates hypercholesterolemia via modulation of gut microbiota and bile acid metabolism. Nat. Commun. 2019, 10, 4971.
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929.
- Liu, T.; Kern, J.T.; Jain, U.; Sonnek, N.M.; Xiong, S.; Simpson, K.F.; VanDussen, K.L.; Winkler, E.S.; Haritunians, T.; Malique, A.; et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe 2021, 29, 988–1001.
- Yan, Y.; Niu, Z.; Sun, C.; Li, P.; Shen, S.; Liu, S.; Wu, Y.; Yun, C.; Jiao, T.; Jia, S.; et al. Hepatic thyroid hormone signalling modulates glucose homeostasis through the regulation of GLP-1 production via bile acid-mediated FXR antagonism. Nat. Commun. 2022, 13, 6408.
- Liu, Y.; Chen, K.; Li, F.; Gu, Z.; Liu, Q.; He, L.; Shao, T.; Song, Q.; Zhu, F.; Zhang, L.; et al. Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice. Hepatology 2020, 71, 2050–2066.
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140.
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177.
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588.
- Münzker, J.; Haase, N.; Till, A.; Sucher, R.; Haange, S.; Nemetschke, L.; Gnad, T.; Jäger, E.; Chen, J.; Riede, S.J.; et al. Functional changes of the gastric bypass microbiota reactivate thermogenic adipose tissue and systemic glucose control via intestinal FXR-TGR5 crosstalk in diet-induced obesity. Microbiome 2022, 10, 96.
- Zhang, S.; Li, R.J.W.; Lim, Y.; Batchuluun, B.; Liu, H.; Waise, T.M.Z.; Lam, T.K.T. FXR in the dorsal vagal complex is sufficient and necessary for upper small intestinal microbiome-mediated changes of TCDCA to alter insulin action in rats. Gut 2021, 70, 1675–1683.
- Xiong, H.; Zhang, C.; Han, L.; Xu, T.; Saeed, K.; Han, J.; Liu, J.; Klaassen, C.D.; Gonzalez, F.J.; Lu, Y.; et al. Suppressed farnesoid X receptor by iron overload in mice and humans potentiates iron-induced hepatotoxicity. Hepatology 2022, 76, 387–403.
- Berrabah, W.; Aumercier, P.; Gheeraert, C.; Dehondt, H.; Bouchaert, E.; Alexandre, J.; Ploton, M.; Mazuy, C.; Caron, S.; Tailleux, A.; et al. Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology 2014, 59, 2022–2033.
- García-Rodríguez, J.L.; Barbier-Torres, L.; Fernández-Álvarez, S.; Gutiérrez-de Juan, V.; Monte, M.J.; Halilbasic, E.; Herranz, D.; Álvarez, L.; Aspichueta, P.; Marín, J.J.G.; et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology 2014, 59, 1972–1983.
- Plass, J.R.M.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.M.; Müller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596.
- Neimark, E.; Chen, F.; Li, X.; Shneider, B.L. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 2004, 40, 149–156.
- Byun, S.; Jung, H.; Chen, J.; Kim, Y.; Kim, D.; Kong, B.; Guo, G.; Kemper, B.; Kemper, J.K. Phosphorylation of hepatic farnesoid X receptor by FGF19 signaling-activated Src maintains cholesterol levels and protects from atherosclerosis. J. Biol. Chem. 2019, 294, 8732–8744.
- De Boer, J.F.; Schonewille, M.; Boesjes, M.; Wolters, H.; Bloks, V.W.; Bos, T.; van Dijk, T.H.; Jurdzinski, A.; Boverhof, R.; Wolters, J.C.; et al. Intestinal Farnesoid X Receptor Controls Transintestinal Cholesterol Excretion in Mice. Gastroenterology 2017, 152, 1126–1138.
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418.
- Hirokane, H.; Nakahara, M.; Tachibana, S.; Shimizu, M.; Sato, R. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J. Biol. Chem. 2004, 279, 45685–45692.
- Claudel, T.; Inoue, Y.; Barbier, O.; Duran-Sandoval, D.; Kosykh, V.; Fruchart, J.; Fruchart, J.; Gonzalez, F.J.; Staels, B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 2003, 125, 544–555.
- Kast, H.R.; Nguyen, C.M.; Sinal, C.J.; Jones, S.A.; Laffitte, B.A.; Reue, K.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: A molecular mechanism linking plasma triglyceride levels to bile acids. Mol. Endocrinol. 2001, 15, 1720–1728.
- Yamagata, K.; Daitoku, H.; Shimamoto, Y.; Matsuzaki, H.; Hirota, K.; Ishida, J.; Fukamizu, A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 2004, 279, 23158–23165.
- Caron, S.; Huaman Samanez, C.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B.; et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211.
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.
- Carino, A.; Biagioli, M.; Marchianò, S.; Scarpelli, P.; Zampella, A.; Limongelli, V.; Fiorucci, S. Disruption of TFGβ-SMAD3 pathway by the nuclear receptor SHP mediates the antifibrotic activities of BAR704, a novel highly selective FXR ligand. Pharmacol. Res. 2018, 131, 17–31.
- Renga, B.; Mencarelli, A.; Migliorati, M.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-γ by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm. Res. 2011, 60, 577–587.
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011, 13, 729–738.
- Miyata, M.; Sakaida, Y.; Matsuzawa, H.; Yoshinari, K.; Yamazoe, Y. Fibroblast growth factor 19 treatment ameliorates disruption of hepatic lipid metabolism in farnesoid X receptor (Fxr)-null mice. Biol. Pharm. Bull. 2011, 34, 1885–1889.
- Bhatnagar, S.; Damron, H.A.; Hillgartner, F.B. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 2009, 284, 10023–10033.
- Marcelin, G.; Jo, Y.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.; Blouet, C.; Chang, J.K.; Chua, S.J. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28.
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.
- Somm, E.; Henry, H.; Bruce, S.J.; Aeby, S.; Rosikiewicz, M.; Sykiotis, G.P.; Asrih, M.; Jornayvaz, F.R.; Denechaud, P.D.; Albrecht, U.; et al. β-Klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue. JCI Insight 2017, 2, e91809.
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia and NAFLD Short title: Bile acids in meta-inflammatory disorders. Gastroenterology 2017, 152, 1679–1694.
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731.
- Makki, K.; Brolin, H.; Petersen, N.; Henricsson, M.; Christensen, D.P.; Khan, M.T.; Wahlström, A.; Bergh, P.; Tremaroli, V.; Schoonjans, K.; et al. 6α-hydroxylated bile acids mediate TGR5 signalling to improve glucose metabolism upon dietary fiber supplementation in mice. Gut 2023, 72, 314–324.
- Wu, Q.; Liang, X.; Wang, K.; Lin, J.; Wang, X.; Wang, P.; Zhang, Y.; Nie, Q.; Liu, H.; Zhang, Z.; et al. Intestinal hypoxia-inducible factor 2α regulates lactate levels to shape the gut microbiome and alter thermogenesis. Cell Metab. 2021, 33, 1988–2003.
- Anhê, F.F.; Nachbar, R.T.; Varin, T.V.; Trottier, J.; Dudonné, S.; Le Barz, M.; Feutry, P.; Pilon, G.; Barbier, O.; Desjardins, Y.; et al. Treatment with camu camu (Myrciaria dubia) prevents obesity by altering the gut microbiota and increasing energy expenditure in diet-induced obese mice. Gut 2019, 68, 453–464.
- Zhang, J.; Ni, Y.; Qian, L.; Fang, Q.; Zheng, T.; Zhang, M.; Gao, Q.; Zhang, Y.; Ni, J.; Hou, X.; et al. Decreased Abundance of Akkermansia muciniphila Leads to the Impairment of Insulin Secretion and Glucose Homeostasis in Lean Type 2 Diabetes. Adv. Sci. 2021, 8, e2100536.
- Zhai, H.; Li, Z.; Peng, M.; Huang, Z.; Qin, T.; Chen, L.; Li, H.; Zhang, H.; Zhang, W.; Xu, G. Takeda G Protein-Coupled Receptor 5-Mechanistic Target of Rapamycin Complex 1 Signaling Contributes to the Increment of Glucagon-Like Peptide-1 Production after Roux-en-Y Gastric Bypass. EBioMedicine 2018, 32, 201–214.
- Perino, A.; Pols, T.W.H.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J. Clin. Investig. 2014, 124, 5424–5436.
- Trabelsi, M.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629.
- Ducastel, S.; Touche, V.; Trabelsi, M.; Boulinguiez, A.; Butruille, L.; Nawrot, M.; Peschard, S.; Chávez-Talavera, O.; Dorchies, E.; Vallez, E.; et al. The nuclear receptor FXR inhibits Glucagon-Like Peptide-1 secretion in response to microbiota-derived Short-Chain Fatty Acids. Sci. Rep. 2020, 10, 174.
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245.
- Wang, Y.; Chen, W.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432.
- Lang, S.; Schnabl, B. Microbiota and Fatty Liver Disease-the Known, the Unknown, and the Future. Cell Host Microbe 2020, 28, 233–244.
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285.
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 740–743.
- Eslamparast, T.; Poustchi, H.; Zamani, F.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled pilot study. Am. J. Clin. Nutr. 2014, 99, 535–542.
- Manzhalii, E.; Virchenko, O.; Falalyeyeva, T.; Beregova, T.; Stremmel, W. Treatment efficacy of a probiotic preparation for non-alcoholic steatohepatitis: A pilot trial. J. Dig. Dis. 2017, 18, 698–703.
- Mofidi, F.; Poustchi, H.; Yari, Z.; Nourinayyer, B.; Merat, S.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in lean patients with non-alcoholic fatty liver disease: A pilot, randomised, double-blind, placebo-controlled, clinical trial. Br. J. Nutr. 2017, 117, 662–668.
- Sayari, S.; Neishaboori, H.; Jameshorani, M. Combined effects of synbiotic and sitagliptin versus sitagliptin alone in patients with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2018, 24, 331–338.
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clément, K. Nonalcoholic Fatty Liver Disease: Modulating Gut Microbiota to Improve Severity? Gastroenterology 2020, 158, 1881–1898.
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511.
- Lindor, K.D.; Kowdley, K.V.; Heathcote, E.J.; Harrison, M.E.; Jorgensen, R.; Angulo, P.; Lymp, J.F.; Burgart, L.; Colin, P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: Results of a randomized trial. Hepatology 2004, 39, 770–778.
- Leuschner, U.F.H.; Lindenthal, B.; Herrmann, G.; Arnold, J.C.; Rössle, M.; Cordes, H.; Zeuzem, S.; Hein, J.; Berg, T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: A double-blind, randomized, placebo-controlled trial. Hepatology 2010, 52, 472–479.
- Traussnigg, S.; Schattenberg, J.M.; Demir, M.; Wiegand, J.; Geier, A.; Teuber, G.; Hofmann, W.P.; Kremer, A.E.; Spreda, F.; Kluwe, J.; et al. Norursodeoxycholic acid versus placebo in the treatment of non-alcoholic fatty liver disease: A double-blind, randomised, placebo-controlled, phase 2 dose-finding trial. Hepatology 2019, 4, 781–793.
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.R.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 2016, 8, 122r–357r.
- Palmer, M.; Jennings, L.; Silberg, D.G.; Bliss, C.; Martin, P. A randomised, double-blind, placebo-controlled phase 1 study of the safety, tolerability and pharmacodynamics of volixibat in overweight and obese but otherwise healthy adults: Implications for treatment of non-alcoholic steatohepatitis. BMC Pharmacol. Toxicol. 2018, 19, 10.
- Newsome, P.N.; Palmer, M.; Freilich, B.; Sheikh, M.Y.; Sheikh, A.; Sarles, H.; Herring, R.; Mantry, P.; Kayali, Z.; Hassanein, T.; et al. Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J. Hepatol. 2020, 73, 231–240.
- Hameed, B.; Terrault, N.A.; Gill, R.M.; Loomba, R.; Chalasani, N.; Hoofnagle, J.H.; Van Natta, M.L. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2018, 47, 645–656.
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965.
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.S.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71.
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643.
- Harrison, S.A.; Bashir, M.R.; Lee, K.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 25–33.
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33.
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60.
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707.
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185.
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J. Safety, Pharmacokinetics, and Pharmacodynamic Effects of a Selective TGR5 Agonist, SB-756050, in Type 2 Diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222.
- Desai, M.S.; Shabier, Z.; Taylor, M.; Lam, F.; Thevananther, S.; Kosters, A.; Karpen, S.J. Hypertrophic cardiomyopathy and dysregulation of cardiac energetics in a mouse model of biliary fibrosis. Hepatology 2010, 51, 2097–2107.
- Fryer, R.M.; Ng, K.J.; Nodop Mazurek, S.G.; Patnaude, L.; Skow, D.J.; Muthukumarana, A.; Gilpin, K.E.; Dinallo, R.M.; Kuzmich, D.; Lord, J.; et al. G protein-coupled bile acid receptor 1 stimulation mediates arterial vasodilation through a K(Ca)1.1 (BK(Ca))-dependent mechanism. J. Pharmacol. Exp. Ther. 2014, 348, 421–431.
- Briere, D.A.; Ruan, X.; Cheng, C.C.; Siesky, A.M.; Fitch, T.E.; Dominguez, C.; Sanfeliciano, S.G.; Montero, C.; Suen, C.S.; Xu, Y.; et al. Novel Small Molecule Agonist of TGR5 Possesses Anti-Diabetic Effects but Causes Gallbladder Filling in Mice. PLoS ONE 2015, 10, e136873.
- Pols, T.W.H.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757.
- Chen, J.; Thomsen, M.; Vitetta, L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J. Cell. Biochem. 2019, 120, 2713–2720.
More