Aging induces several stress response pathways to counterbalance detrimental changes associated with this process. These pathways include nutrient signaling, proteostasis, mitochondrial quality control and DNA damage response. At the cellular level, these pathways are controlled by evolutionarily conserved signaling molecules, such as 5'’AMP-activated protein kinase (AMPK), mechanistic target of rapamycin (mTOR), insulin/insulin-like growth factor 1 (IGF-1) and sirtuins, including SIRT1. Peroxisome proliferation-activated receptor coactivator 1 alpha (PGC-1α), encoded by the PPARGC1A gene, playing an important role in antioxidant defense and mitochondrial biogenesis, may interact with these molecules influencing lifespan and general fitness. Perturbation in the aging stress response may lead to aging-related disorders, including age-related macular degeneration (AMD), the main reason for vision loss in the elderly. This is supported by studies showing an important role of disturbances in mitochondrial metabolism, DDR and autophagy in AMD pathogenesis. In addition, disturbed expression of PGC-1α was shown to associate with AMD. Therefore, the aging stress response may be critical for AMD pathogenesis, and further studies are needed to precisely determine mechanisms underlying its role in AMD.
- the aging stress response
- aging
- age-related macular degeneration
- AMD
- the aging stress response,aging,age-related macular degeneration,AMD
Please note: Below is an entry draft based on your previous paper, which is wrirren tightly around the entry title. Since it may not be very comprehensive, we kindly invite you to modify it (both title and content can be replaced) according to your extensive expertise. We believe this entry would be beneficial to generate more views for your work. In addition, no worry about the entry format, we will correct it and add references after the entry is online (you can also send a word file to us, and we will help you with submitting).
1. Introduction
The biological fitness of the human organism is supported by the phylogenetically conserved stress response, maintenance and repair pathways. They include nutrients and energy signaling, proteostasis, DNA damage response (DDR), mitochondrial quality control (mtQC) and enzymes metabolizing toxic species [1]. The efficacy of these pathways declines with aging, but the process of aging is also associated with diverse stresses that impose signals coming from the environment (reviewed in [2]). Therefore, an aging organism faces increased stress, which should be counterbalanced by the aging stress response with signaling pathways to maintain cellular and organismal homeostasis (reviewed in [3]). The impairment of the aging stress response may contribute to syndromes associated with premature aging, age-related diseases and cancer.
Age-related macular degeneration (AMD) is the primary cause of legal blindness in the elderly in developed countries. Its estimated prevalence in the world is 196 million in 2020 and is projected to increase to 288 million by 2040. Despite these numbers, no effective treatment is available, except for some specific cases of the neovascular (wet) form of AMD (reviewed in [4]). However, the treatment in those cases is mainly aimed at vision loss prevention. This limitation of effective therapeutic options is likely due to highly incomplete knowledge of mechanism(s) involved in AMD pathogenesis. It is commonly accepted that aging is the most serious risk factor in AMD. Therefore, aging-related stress and response to this stress may contribute to AMD pathogenesis. Oxidative stress is often considered a major risk factor in AMD [5]. It is associated with increased levels of reactive oxygen and nitrogen species (ROS and RNS) that may damage cellular structures and macromolecules, including proteins and DNA. This may result in misfolded and dysfunctional proteins and consequently dysfunctional subcellular structures and organelles. These proteins should be removed and recycled by proteostasis, which dynamically regulates the proteome to keep it balanced and functional and is an essential pathway of the stress response [6]. Damage to DNA induces DDR, another stress response component [7]. However, oxidative stress and increased ROS/RNS levels are associated with several other AMD risk factors, including improper diet, tobacco smoking, blue light exposure and others [8]. Moreover, oxidative stress is common in the retina as it is the tissue of high consumption of oxygen, contains a high proportion of polyunsaturated fatty acids and is exposed to visible light. Therefore, oxidative stress is also present in young retinas that are generally not susceptible to AMD, and there must be an additional factor(s) associated with age that superimposes the stress in older individuals. There are substantial differences in everyday life between young and older adults, such as nutrient intake and physical activity that may influence AMD susceptibility [9]. The aging stress response seems to be a prime candidate to distinguish AMD susceptibility of younger and older adults as it combats many AMD risk factors and is declined with aging.
2. The Aging Stress Response
Aging is frequently understood as the process resulting from the accumulation of detrimental biological changes over time, which increase an organism'’s susceptibility to disease and make it more likely to die. However, the causal relationship between the biological changes that occur with time (biological aging) and metrical (chronological) aging is far from understanding [10].
Aging may be considered as a regulated process controlled by some conserved signaling pathways, including the insulin/IGF-1 (insulin-like growth factor 1), mTOR (mechanistic target of rapamycin), AMPK (5'’AMP-activated protein kinase) and sirtuins [11]. These pathways detect stress-related events and activate proteins of adaptive cellular responses that reduce the consequences of environmental insults and aging-related stress [3]. These adaptive responses include DNA repair, proteostasis with autophagy and mitochondrial quality control—all of them have been reported impaired in AMD patients and AMD cellular and animal models [12][13][14][15][16][12,13,14,15,16].
2.1. Nutrient-Signaling Pathways in Aging
Nutrient signaling plays an important role in the control of aging and age-related disorders, and it was the first pathway to be demonstrated to do so in model organisms [17]. Caloric restriction, one of the best-known way of lifespan extension, acts through multiple signaling pathways, including the insulin/IGF-1, mTOR and AMPK pathways and sirtuins [18].
In an aging organism, the level of active IGF-1 decreases, which has been associated with human frailty and cognitive decline [19]. Mutations in the genes of the insulin/IGF-1 pathway are reported to prolong the life of model organisms [20]. This pathway is a molecular connection between dietary intake and the cellular stress response pathway, so it can be modulated by a dietary intervention. Aging and age-related pathologies relate to the metabolic status of sirtuins, a family of highly conserved proteins [21][22][21,22]. They display NAD+ (nicotinamide adenine dinucleotide)-dependent protein deacetylase activity, and this is NAD+-sensing that determines their association with metabolic status [23]. Among seven mammalian sirtuins, SIRT1 is regulated by nutrient status in the cell, triggers stress response pathways and induces changes in energy metabolism [24]. The expression and activity of SIRT1 decrease with age and a diet rich in fat [25]. On the other hand, its activity increases in nutrients deficiency, including caloric restriction [26]. Active SIRT1 deacetylases several substrates that are involved in aging and stress response, including peroxisome proliferation-activated receptor coactivator 1 alpha (PGC-1α) [27]. Numerous studies suggest that SIRT1 may be involved in the extension of lifespan in stress conditions and can do so through the regulation of stress–response processes, including DNA repair, mitochondria and protein quality control as well as cell survival [21] (Figure 1). It is still an open question whether it can also do so in normal conditions.
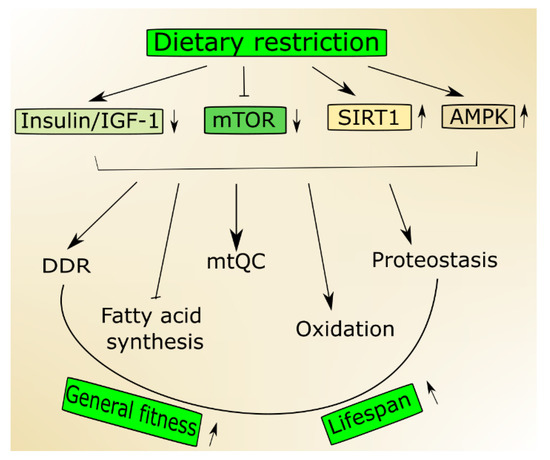
Figure 1. Nutrition signaling includes proteins and signaling pathways important in aging. Dietary restriction activates 5'AMP-activated protein kinase (AMPK) and sirtuin 1 (SIRT1) but inhibits the insulin growth factor 1 (insulin/IGF-1) and mechanistic target of rapamycin (mTOR) that stimulate genome maintenance by improving DNA damage response (DDR) and mitochondrial functions and eliminate damaged mitochondria through mitochondrial quality control (mtQC). Both mTOR decrease and SIRT1 increase may contribute to improved proteostasis, and AMPK supports the change from fatty acid synthesis to oxidation. Altogether these actions may result in an increase of biological fitness and extension of the lifespan of an organism.
A low status of cellular energy, which can be reflected by a low ATP/AMP ratio, may activate AMPK that is not only a sensor of such status but also becomes an energy reservoir [28]. The stress of a low-energy state evokes an increase in catabolic pathways promoted by signaling pathways transcriptionally and post-transcriptionally activated by AMPK [29][30][29,30]. AMPK promotes the switch from fatty acid synthesis to oxidation during nutritional stress resulting in low ATP level [3].
Another nutrient sensor, mTOR, integrates environmental signals, including those coming from nutrients, to control growth and metabolism in eukaryotic cells [31]. Nutrient limitation decreases mTOR, and its activity is suppressed in models of longevity (reviewed in [32]). Moreover, its inhibition extended lifespan in various model organisms [33]. In mammals, mTOR acts in two separate signaling complexes, mTORC1 and mTORC2 and their age-related effects are mainly underlined by modulation of protein synthesis and autophagy [34].
2.2. Mitochondria in Aging
Although the mitochondrial theory of aging, originating from Harman'’s "“free radical theory of aging"”, is no longer a paradigm, mitochondria are still a central element in the process of aging [35]. Such a prominent role is supported by research with overexpression of human catalase targeted to mouse mitochondria and extending lifespan of transgenic animals by about five months as compared with three months or one month for the enzyme targeted the peroxisome or the nucleus, respectively [36]. The signs of aging: cardiac pathology and cataract development were delayed in animals with catalase targeted to mitochondria. Moreover, the most pronounced antioxidant effect was observed for mtDNA, suggesting that aging may be linked with the production of ROS in mitochondria [37]. Mice with human catalase targeted to mitochondria were also exploited in models of various diseases, including metabolic syndrome and atherosclerosis, cardiac aging, heart failure, skeletal muscle pathology, sensory defect, neurodegenerative diseases and cancer, suggesting that mitochondrial production of ROS and damage to mitochondria may play an essential role in the process of aging and aging-related pathologies [38].
Studies on other mouse models, including animals with mutations in the gene encoding polymerase gamma, the mitochondrial replicase, showed that the association between aging and mitochondrial ROS production and mitochondrial homeostasis was complex [39][40][41][39,40,41]. These studies also pointed to the role of apoptosis in aging. Mutations in mtDNA may either extend or reduce the lifespan of model organisms, but the lack of mtDNA in yeast ("“the petit mutation"”) is linked with lifespan extension [42]. As was later shown, this effect was attributed to the lack of mtDNA per se and not to changes in respiration and reduced ROS production [43].
Mitochondrial quality control is accomplished by the coordination of mitochondrial biogenesis, dynamics and mitophagy, a specialized pathway of autophagy [44] (Figure 2). Mitochondrial transcription factors, including mitochondrial transcription factor A (TFAM), are encoded by nuclear genes and transported to mitochondria to target mtDNA. TFAM upregulates genes encoding the mitochondrial electron transport chain (mtETC), resulting in an elevated oxygen intake, ATP production and mitochondrial content. Mitochondrial turnover is mainly controlled by fusion (mitofusin 1 (MFN1), MFN2 and mitochondrial dynamin-like 120 kDa protein (OPA1)) and fission (dynamin 1-like protein (DNM1L) and mitochondrial fission 1 protein (FIS1)) proteins [45][46][45,46]. Damaged mitochondrial components can be degraded and recycled by mitophagy [47].
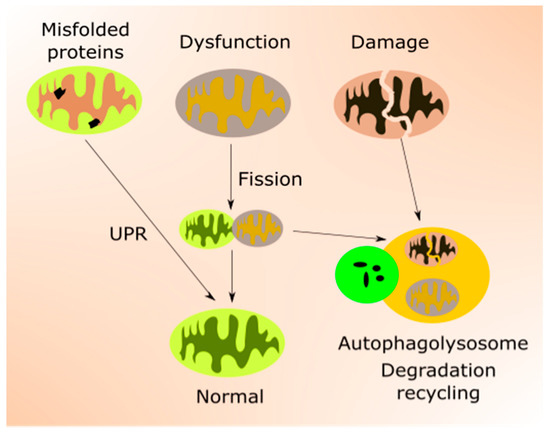
Figure 2.
Several pathways and proteins control mitochondrial response to stress associated with low energy allowing mitochondria to adjust their energy usage and the number of organelles [3]. These include PGC-1α, sirtuins, mTOR and AMPK. Therefore, mitochondrial response to age-related stress crosstalk with nutrition signaling. PGC-1α is a key regulator of mitochondrial biogenesis and an important factor in other aspects of mtQC [42]. Moreover, emerging evidence suggests that PGC-1α is a major regulator of lifespan and its interaction with SIRT1, AMPK, and mTOR is important for this function [48][49][50][51][48,49,50,51].
A mild mitochondrial impairment may activate a stress response pathway, which promotes a protective milieu counterbalancing a potentially shorter lifespan associated with the mitochondrial impairment [3]. Whether it is a classical hormesis response or results from the interaction of PGC-1α with SIRT1, AMPK and mTOR, remains to be established, although mTOR seems to be a prime candidate to link mitochondrial biogenesis and cellular metabolism with lifespan [3].
2.3. DDR in Aging
Increasing DNA damage resulting from the aging process is predicted by all theories of aging [52]. Moreover, not only DNA but other cellular macromolecules undergo damage in aging [53]. Unrepaired DNA damage may be changed into mutation (Figure 3). Therefore, aging is associated with accumulating damage to cellular molecules that may become non-functional. This may contribute to age-related degenerative diseases, and increased levels of DNA damage and mutations lead to genomic instability, associated with almost all, if not all, cancers [54][55][56][54,55,56]. In addition, the epigenetic profile changes with aging, which reflects a complex relationship between aging and epigenetics (reviewed in [57]). Many aspects of the nutritional signaling in aging, including longevity-related interactions of AMPK, mTOR, SIRT1 and PGC-1α, are underlined by changes in the epigenetic profile [58].
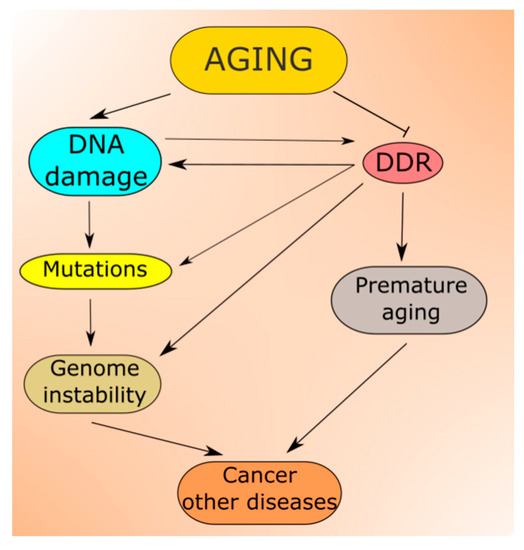
Figure 3.
Aging-related DNA damage induces DDR, which may be impaired from the very start as potentially each of its proteins can be damaged in aging. In fact, many reports suggest that the capacity of DNA repair, the main component of DDR, declines with age, which contributes to both increased DNA damage and genomic instability [52][59][60][61][62][52,59,60,61,62].
Although we are far from the generally accepted definition of aging, premature aging is likely commonly understood. In humans, premature aging and early death are associated with defects in DNA repair or, more generally—in DDR [63][64][65][66][67][63,64,65,66,67]. However, several studies report not only an association but also a causal relationship between DNA repair defects and premature aging [68][69][70][71][72][68,69,70,71,72]. Werner syndrome is likely the most characterized human premature aging disorder, which is closely associated with defects in DNA repair as this syndrome is caused by recessive mutations in the RecQ gene, encoding a DNA helicase involved in DNA repair [73].
Studies of Werner syndrome and other human progeroid disorders suggest an association between DDR and signaling pathways involved in the aging process [3]. Mutations in the essential DNA repair genes XPF-ERCC1 (ERCC4 excision repair 4, endonuclease catalytic subunit) and TP53 (tumor protein p53) resulted in downregulation of the insulin/IGF pathway in cells from progeroid syndrome patients and mice [74][75][74,75].
Another example of the association of DDR with aging is ataxia–telangiectasia (AT), underlined by mutations in the ATM (ATM serine/threonine kinase, ataxia telangiectasia mutated) gene [76]. The ATM protein, along with ATR (ATR serine/threonine kinase, ataxia telangiectasia and Rad3-related protein), is the central regulator of DDR, responsible for the transduction of the signal coming from DDR sensors, recognizing damaged DNA or disturbed DNA replication and activating DDR effectors, controlling cellular processes aimed at restoring the DNA structure or resuming DNA replication [77]. In general, ATM and ATR have a broad spectrum of substrates, including proteins playing an important role in signaling pathways regulating aging [78].
DNA damage response may not only restore the integrity and fidelity of DNA, but it may induce senescence or programmed cell death, which may contribute to aging phenotype [79]. Moreover, some DNA damages may be tolerated, depending on the cellular context. In general, when DNA damage does not exceed the cell repair capacity, it is repaired, but when the extent of the damage is above such capacity, the cell cycle is arrested for giving the cell additional time for repair. If this fails, the cell becomes senescent or is directed to a programmable death pathway, usually apoptosis. Therefore, DDR may differ in mitotic and postmitotic cells, and this is likely responsible for different AT phenotypes in different tissues: loss of ATM-mediated signaling in mitotic cells results in cancer(s), but in postmitotic neurons—in degeneration [80][81][80,81]. In addition, in mitotic cells, DDR may tolerate or misrepair some DNA damages to preserve the process of replication. Therefore, genotoxic stress induces a response with many pathways that may be affected by aging, leading to enhancing aging-related phenotypes, including aging-related disease and death.
DNA damage response in mitochondria (mtDDR), although not fully known, is likely poorer than its nuclear counterpart. mtDDR does not directly induce apoptosis but may lead to the degradation of highly damaged mtDNA. Several aspects of aging-related mtDNA are directly or indirectly associated with mechanisms of mtQC, described in the previous section.
2.4. Autophagy and Aging
Proteostasis, a process ensuring proper protein folding, sequestering of misfolded proteins, degradation of damaged or no longer needed proteins, is performed by the collective action of a set of molecular chaperones, transcription factors and cofactors.
Proper protein folding is ensured by UPR, improperly folded proteins are sequestered in inclusion bodies, and degradation occurs in the proteasome and in autophagy (Figure 4). Proteostasis is declined with age, which may result in the escape of misfolded proteins from sequestration and non-functional/damaged proteins from degradation [82]. Both misfolded and damaged proteins may be dysfunctional, further contributing to deleterious changes, increasing susceptibility of the organism to disease, and making it more likely to die.
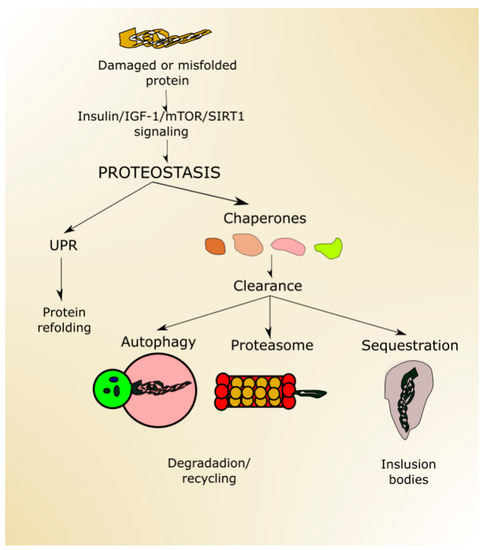
Figure 4.
Autophagy declines with aging, contributing to the accumulation of dysfunctional organelles and proteins as well as their aggregation [83]. Conversely, efficient autophagy may contribute to lifespan extension, as it was shown in research on model organisms (reviewed in [84]). However, few studies show that autophagy alone is sufficient to extend lifespan—instead, it is required for life extension induced by caloric restriction or reduced insulin signaling [85]. It seems that autophagy may take a special position in proteostasis as it may be involved in many effects, phenomena and systems, including oxidative stress defense, DDR and perform the pro-death and pro-survival functions [86]. Therefore, autophagy may contribute to the regulation of lifespan on many distinct pathways.
Mammalian studies on autophagy confirm research on lower organisms and demonstrate an essential role of autophagy in aging and age-related neurodegeneration [87]. Moreover, autophagy, due to its multifaceted nature, may play a role in many aspects of age-related stress responses. Autophagy activation is closely associated with nutrient status and various other stimuli, such as energy stress, endoplasmic reticulum (ER) stress, pathogen- and danger-associated molecular patterns, hypoxia, redox stress and mitochondrial damage [88]. During starvation, autophagy provides cell recycled amino acids for protein synthesis.
Osteoarthritis is the most common form of arthritis and can be considered as a sign of aging because aging is its primary risk factor [89]. It was shown that SIRT1 was decreased in human normal aged and osteoarthritis cartilage compared with young cartilage [90]. Moreover, activation of SIRT1 increased autophagy in chondrocytes by the deacetylation of essential autophagy proteins, including Beclin1, ATG5 (autophagy-related 5), ATG7, LC3 (MAP1LC3A, microtubule-associated protein 1 light chain 3 alpha) in an mTOR/ULK1 (Unc-51-like autophagy activating kinase 1)-independent manner. These results confirm an important role of SIRT1 in aging-related stress response through the involvement in the autophagic pathway.
Mitochondria-specific autophagy, or mitophagy, also declines with age, and its dysfunction may further potentiate an age-related phenotype [91]. In line with this statement is the observation that old mice fed with a mitophagy inducer showed an improvement in age-related cardiac functions and increased levels of mtQC proteins, including PARKIN, BNIP3 (BCL2 interacting protein 3), SIRT3 and PGC-1α [92].
3. Age-Related Macular Degeneration—A Disease of Aging Stress Response
AMD affects the macula, a small structure in the center of the retina, responsible for sharp and color vision.
Clinically, advanced AMD may be divided into two forms: dry (atrophic) and wet (neovascular) AMD (Figure 5). Dry AMD is characterized by the presence of drusen, small white or yellowish objects that are made of protein and lipids. Dry AMD may lead to vision loss as the development of this disease to geographic atrophy (GA) may impair the functionality of photoreceptors and eventually cause their death along with underlying retinal pigment epithelium (RPE) cells atrophy [93][94][93,94]. In wet AMD, the RPE induces the synthesis of angiogenic factors that stimulate the production of new blood vessels from supporting choriocapillaris (choroidal neovascularization, CNV). These vessels penetrate the Bruch'’s membrane-RPE complex leading to photoreceptor death. In some cases, large hemorrhages develop in the subretinal area.
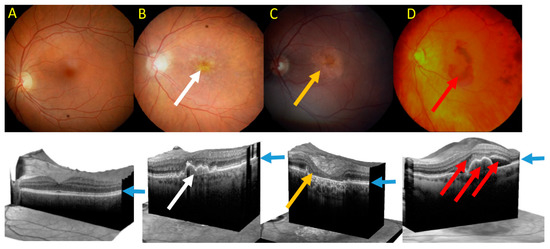
Figure 5.
A
B
C
D
Although AMD pathogenesis is not fully known, many factors, both genetic/epigenetic and environmental/lifestyle, may be involved [95]. Mechanisms and factors of AMD pathogenesis are described in many excellent reviews (for example, [96][97][98][99][96,97,98,99]). In this review, we focus on the elements of AMD pathophysiology directly related to aging and the aging stress response: autophagy as a pathway of proteostasis, DDR and mtQC.
As mentioned, aging is likely the most serious risk factor for AMD, and it induces disturbances in signaling pathways, which are important in AMD. These changes can be potentiated by other environmental/lifestyle factors. For instance, aging evokes detrimental changes in mitochondria that may lead to ROS overproduction, damage to mtDNA and synthesis of faulty proteins of mtETC complex [100]. Damaged mtETC produces more ROS than in normal conditions. However, a similar effect can be obtained by environmental factors damaging mtDNA. This strong dependence of AMD pathogenesis on aging is supported by the presence of increased levels of amyloid in AMD retinas, suggesting that AMD may be a kind of dementia of the eye [101][102][101,102]. Many studies report an enhanced release of the brain amyloid beta 1–42 (Aβ) in stress conditions [103]. Moreover, the production and clearance of Aβ are reported to depend on stress [104]. Therefore, the deposition of Aβ may result from a decline of the stress response with aging. In summary, amyloid beta deposits observed in AMD retinas may be associated with a declined aging stress response.
To date, more than half of the AMD genomic heritability can be explained by both common and rare genetic variants in 34 genetic loci, which are mostly involved in the complement system, extracellular matrix remodeling and lipid metabolism [105]. A common variant (rs1061170) in the complement factor H (CFH) has been identified by several research groups to strongly influence AMD risk [106]. The age-related maculopathy susceptibility 2 and high-temperature requirement A serine peptidase 1 (ARMS2/HTRA1) is another locus reported to be strongly involved in AMD pathogenesis [107].