In chronic kidney disease (CKD), the kidneys gradually lose their ability to excrete toxic waste into the urine, leading to a buildup of serum uremic toxins in the body
[1]. CKD patients may experience higher fracture and mortality rates, associated with CKD–mineral bone disease (CKD–MBD), characterized by abnormal bone turnover and reduced mineralization and volume
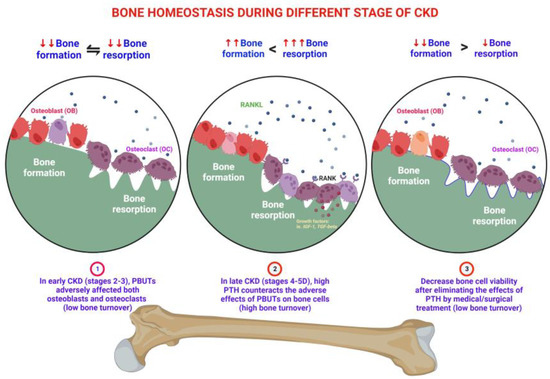
[2]. In patients with early CKD (stages 2–3), wingless (Wnt) signaling inhibitors, such as Dickkopf-1 (DKK1), sclerostin (SOST), and secreted frizzled-related protein (sFRP), are secreted from the kidney or osteocytes in bone or calcified soft tissue. These inhibitors can affect osteoblasts, leading to a decrease in their viability. Additionally, the retention of protein-bound uremic toxins (PBUTs) such as indoxyl sulfate (IS)/p-cresyl sulfate (pCS) further impairs the viability and function of both osteoblasts and osteoclasts
[3]. CKD leads to alterations in the gut microbiome (dysbiosis) and the accumulation of gut-derived uremic toxins in plasma
[4]. It has previously been shown that the uremic toxins IS and pCS are inversely correlated to the estimated glomerular filtration rate (eGFR) across all stages of CKD
[5][6][5,6]. IS is associated with low bone turnover. It suppresses bone formation by downregulating the parathyroid hormone receptor (PTHR)
[7], inhibits Wnt signaling
[8], and promotes apoptosis
[9] in cultured osteoblast cells. The administration of an intestinal adsorbent, which reduces uremic toxins, increases bone turnover in uremic rats
[10]. The term “uremic osteoporosis” is used to describe the loss of bone quality with normal bone mass due to the effects of PBUTs on bone health
[3]. When renal function progressively deteriorates, metabolic acidosis and hyponatremia contribute to bone loss
[11]. In patients with CKD stages 4–5, the dysregulation of calcium, phosphate, vitamin D, and parathyroid hormone (PTH) can lead to varying levels of PTH
[12]. High-turnover bone disease develops (CKD stages 4–5) when high serum PTH levels overcome peripheral PTH resistance and other inhibitory factors of bone formation
[13]. High levels of PTH in progressive CKD upregulate the receptor activator of nuclear factor-kappa B ligand (RANKL) mRNA and inhibit osteoprotegerin (OPG) gene expression in bone marrow stromal osteoblasts
[14], leading to increased quantities of osteoclasts and osteoblasts
[15]. High PTH levels drive the indolent OB into high viability and function, but poor quality in behavior, resulting in both bone quality and quantity loss
[13]. However, with the medical or surgical treatment of secondary hyperparathyroidism (SHPT), which involves the removal of the stimulator of PTH, the bone cells may return to the innate low bone cell viability status, the low bone turnover disorders
[12] (
Figure 1).
2. Pathophysiology of PBUTs: IS and pCS
PBUTs accumulate in CKD patients due to impaired renal clearance. These solutes, which are normally excreted by healthy kidneys, accumulate in CKD due to reduced filtration, impaired tubular secretion, harmful gut microbial metabolism, and unfavorable dietary precursor
[33]. PBUTs, such as IS and pCS, tightly bind to proteins such as serum albumin, making their removal from the body challenging
[34].
3. Effects of PBUTs on Bone
3.1. PBUTs Influence Bone Metabolism
Exposure to PBUTs associated with CKD and a sedentary lifestyle can impact bone metabolism. These factors can contribute to the conversion of white fat cells into brown fat cells, the recruitment of immune cells, and the release of proinflammatory cytokines, leading to chronic inflammation
[34]. However, engaging in physical activity can counteract these effects by inducing sarcomeric contracture in muscles, which releases anti-inflammatory muscle factors
[35][88]. Various myokines, such as IL6, irisin, IGF-1, BDNF (brain-derived neurotrophic factor), FGF2, and myostatin, can have either anabolic or catabolic effects on bones. Additionally, osteocalcin enhances muscle anabolism, while sclerostin attenuates muscle catabolism
[36][89]. The browning of adipocytes stimulates thermogenesis through the release of lipolytic myokines, and the activation of adipokines, such as TNF-α, adiponectin, resistin, and leptin, can modulate muscle and bone metabolism
[37][90]. The interactions between PBUTs, physical activity, myokines, and adipokines influence the complex relationship between bone health and overall metabolism.
4. IS and pCS on the Muscle
In catabolic diseases such as CKD, the presence of uremic toxins affects the muscle. In CKD, elevated levels of myostatin, TGF-β, and glucocorticoids contribute to increased expression of autophagy-related genes, atrogin-1, and muscle RING-finger 1 (MuRF1). As a result, this leads to a reduced activation of the insulin/IGF-1-Akt-mTOR pathway, resulting in diminished protein synthesis and muscle mass
[38][117].
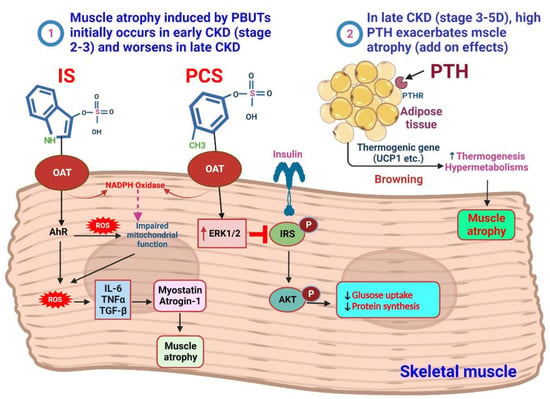
IS, which enters muscle cells through OATs, plays a role in stimulating the AhR and NADPH oxidase pathways
[39][118]. This stimulation leads to the production of ROS. The excessive ROS production, combined with the overexpression of myostatin and atrogin-1, by triggering the production of inflammatory cytokines, IS contributes to muscle atrophy. Furthermore, ROS production negatively affects mitochondrial function, further impacting muscle health
[40][41][119,120]. pCS, on the other hand, induces insulin resistance in muscles by reducing the capacity of insulin or IGF-1-IRS-AKT signaling through increased activity of ERK1/2. This interference with insulin signaling can impair protein synthesis and muscle growth
[42][43][121,122]. Understanding these effects is crucial for developing targeted interventions to preserve muscle mass and function in patients with catabolic conditions (
Figure 2).
Figure 2. Mechanism of PBUTs and PTH-mediated skeletal muscle atrophy. Redox signaling changes in CKD, caused by uremic toxins, inflammation, and metabolic/hormonal shifts, resulting in oxidative stress, leading to muscle wasting and bone loss. ① Mild accumulation of PBUTs significantly contributes to muscle wasting in early-stage CKD (stages 2–3). Molecular mechanisms involved include imbalanced protein degradation and synthesis, increased reactive ROS and inflammatory cytokines, activation of myostatin and atrogene expression, impaired mitochondrial function, and negative effects from uremic toxins, parathyroid hormone, glucocorticoids, and angiotensin II. IS enters muscle cells using an OAT, where IS stimulates the pathway of AhR and NADPH oxidase to induce the production of ROS. Excessive production of ROS will produce inflammatory cytokines when myostatin and atrogin-1 are overexpressed, and this will be linked to muscle atrophy. The production of ROS has an impact on mitochondrial function. pCS induces insulin resistance in muscles due to decreased insulin or IGF-1-IRS-AKT capacity triggered by ERK1/2 activity. In late CKD (stages 4–5D), cachexia is a syndrome characterized by further increased energy expenditure and loss of muscle and adipose tissues. In addition to the further increase in PBUT levels, the excessive levels of PTH have detrimental effects on skeletal muscle metabolism, leading to aggravated muscle weakness and atrophy. ② High PTH levels are associated with sarcopenia and contribute to muscle loss. PTH and PTHrP drive adipose tissue browning and muscle wasting in cachexia. Elevated PTH indirectly leads to decreased muscle protein synthesis by acting on PTH receptors expressed in adipose tissue, thereby activating the expression of thermogenic genes, and finally causing muscle hypermetabolism and atrophy.
5. Crosslinks between Bones and Muscles
Bone health relies on the coordinated actions of osteocytes, osteoblasts, and osteoclasts, which secrete bone-derived factors known as osteokines. Aging and metabolic disease can disrupt this process, leading to bone loss and an elevated risk of fractures
[44][142]. Endocrine factors, genetics, and development have been extensively studied in the relationship between osteoporosis and sarcopenia. In particular, the role of myokines and osteokines has gained attention in understanding the pathogenesis of osteopenia.
The Our
esearchers' recent clinical cohort found that ESRD patients had low handgrip strength, possibly due to higher levels of IS
[45][143]. Elevated IS levels are associated with muscle issues, and incorporating AST-120 in CKD treatment shows promise for improving sarcopenia
[46][144]. Another study revealed a positive correlation between IS levels and bone formation rate in predialysis CKD patients
[47][106].
IS levels increase as renal function declines, with ESRD patients having significantly higher levels. According to a previous report, the IS concentrations are as follows: 0.97 μM at stage 1, 1.98 μM at stage 2, 12.73 μM at stage 3, 21.48 μM at stage 4, 78.79 μM (19.8 μg/mL) at stage 5, and 169.12 μM (44.86 μg/mL) at stage 5D
[48][145]. In
theour previous study,
the researchers we investigated the impact of IS on bone cells and their development. Using osteoclast precursor cells and Raw 264.7 cells,
the researchers we treated osteoclast precursor cells with 100 μM of IS, which represents the average concentration found in the serum of patients with end-stage renal disease (ESRD, stage 5D). Additionally,
the researchers cwe cultured Raw 264.7 cells with 50 ng/mL of soluble RANKL in various IS concentrations (0, 20, 100, 250, 500, and 1000 µM) to assess osteoclastogenesis at different IS levels. The results revealed that as the concentration of IS increased beyond 100 µM, there was a dose-dependent decrease in the percentage of TRAP-positive cells and the number of mature osteoclast cells. Furthermore, when the cells were exposed to IS for longer durations (5 days), the suppression of osteoclastogenesis became more pronounced. These findings indicate that the impact of IS on osteoclastogenesis is both concentration dependent and influenced by the duration of exposure
[49][146].
In another study,
thwe
researchers investigated the impact of IS on osteoblast development. Primary osteoblast cells were cultured in an osteogenesis medium and exposed to various concentrations of IS (0, 20, 100, 250, 500, and 1000 μM). The first set of cultures lasted for 14 days, during which
the researchers we observed the effects of IS on osteoblast development using alkaline phosphatase staining, which reflects osteoblast activity. As the concentration of IS increased,
the researcherswe noticed a dose-dependent decrease in alkaline phosphatase activity, indicating impaired osteoblast development in response to higher IS concentrations. The second set of cultures continued for 21 days, and
the researchers ewe examined the effects of IS on osteoblast mineralization using alizarin red staining. Once again,
the researchers we found a dose-dependent decrease in alizarin red staining as the IS concentration increased. This finding suggests that the mineralization capacity of osteoblasts was adversely affected by higher levels of IS. Overall, this
researchstudy demonstrated that increasing concentrations of IS negatively impact osteoblast development and mineralization, highlighting the potential detrimental effects of IS on bone health
[50][147]. However, further investigation is necessary to fully understand the relationship between mineral and bone disorder and its impact on the balance of bone and muscle in CKD patients.
6. Possible Therapeutic Considerations for Bone and Muscular Health in CKD
PBUTs can negatively affect skeletal and muscle functions, leading clinicians to recognize the effectiveness of antiuremic toxin medications. The subsequent text will explore various therapeutic considerations.
Removal or Decrease of PBUT Precursor from the Intestinal Tract
AST-120, an oral spherical carbon adsorbent, absorbs protein-bound uremic toxin precursors from the intestine
[51][162]. In uremic rats, AST-120 shows potential in preventing the decline of low bone turnover
[52][101]. Previous studies have indicated that AST-120, when administered orally along with a low-protein diet, may slow the progression of renal function deterioration
[53][69]. Advanced kidney disease patients often exhibit high levels of IS in the serum
[54][163], but treatment with AST-120 can reduce serum IS levels compared with the control group with no treatment. Exercise capacity in skeletal muscle is positively correlated with mitochondrial function, which is mainly controlled by mitochondrial biosynthesis and degradation
[55][56][164,165]. AST-120 treatment reduces IS levels, restoring mitochondrial function and improving exercise capacity in skeletal muscles
[57][166].
Resveratrol (RSV) is a small polyphenol found in grape skins, cocoa, blueberries, and peanuts
[58][167]. It improves the tight junction of the intestinal epithelium, providing benefits
[59][168]. RSV inhibits hepatic sulfotransferase, reducing the synthesis of IS and offering cardiovascular protection
[60][169]. In lower concentrations, RSV reduces reactive oxygen and nitrogen species formation in primary granulosa cells and promotes mitochondrial biogenesis
[61][62][170,171]. RSV therapy promotes osteogenic differentiation and mitochondrial activity in bone marrow–derived mesenchymal stem cells
[63][172]. Additionally, RSV influences the mitogen-activated protein kinase (MAPK) pathway, contributing to skeletal development
[64][173].
Probiotics, living microorganisms, offer multiple benefits, such as restoring balance to the intestinal microbiota, improving mucosal integrity and cellular tight junctions, and regulating the microbiota environment by lowering pH. Indole and p-cresol, produced by the intestinal microbiota, bind to plasma albumin and are typically excreted in urine
[65][174]. The accumulation of PBUTs in CKD disrupts gut microbiota and harms bone metabolism and cardiovascular health
[65][66][174,175]. PBUTs generate excessive reactive oxygen species that impair osteoblast and osteoclast function and induce PTH resistance in bone cells
[66][175]. Probiotics can mitigate IS toxicity, protect cardiovascular function, and decelerate renal deterioration.
Probenecid, an inhibitor of the organic anion transporter (OAT), demonstrated a dose-dependent blocking effect on IS inhibition in a previous mouse study
[67][93]. IS enters cells via OAT-3 and promotes oxidative stress, leading to alterations in osteoblast function and decreased PTHR expression
[7]. Probenecid protects osteoblasts from IS-induced damage and stabilizes osteoclasts, promoting favorable bone metabolism.
7. Other Possible Therapeutic Strategies
Metabolic acidosis, aluminum toxicity, and diabetes are factors associated with bone and muscle loss. It is important to treat hyperglycemia, avoid aluminum-containing phosphate binders, and correct metabolic acidosis, especially in CKD patients. Ursolic acid has shown promise in improving mitochondrial biosynthesis and mitigating muscle dysfunction caused by CKD by influencing mouse myoblast differentiation, IL-6 secretion, and ATP levels
[68][176]. Furthermore, the reduction of hyperphosphatemia has been associated with decreased inflammation and improvement in anemia and skeletal muscle wasting
[69][177].
A previous study showed that combining omega-3 fatty acid and menaquinone-7 supplementation in uremic rats can help prevent aortic vascular calcification, reduce osteoclast activation in the bone, and improve sarcopenia-related molecules
[70][178]. Nutrition plays a significant role in sarcopenia. A clinical study found that the elderly hemodialysis sarcopenia index (EHSI) can identify sarcopenia diagnosed by the European Working Group on Sarcopenia in Older People (EWGSOP) second meeting using easily accessible anthropometric and nutritional parameters. A combination of gender, age, serum albumin, phosphate, and cholesterol can effectively predict the severity of sarcopenia
[71][179]. The current nutritional therapy for uremic sarcopenia includes oral supplements, parenteral nutrition, enteral nutrition, high protein and fiber diet, and gastrectomy
[72][180].
Nutritional interventions, along with exercise, have been shown to have positive effects on measures of sarcopenia in individuals with CKD. Longer intervention duration, supervision, and participant adherence increase muscle strength outcomes. CKD patients may require higher intensity and progressive loading in resistance exercise to see noticeable results in muscle mass. Although the current evidence for progressive resistance exercise in CKD is encouraging, real-life applications in clinical settings are limited. A multidisciplinary patient-centered approach with regular follow-up may be beneficial for managing the complexity of sarcopenia in CKD
[73][181].
Combining targeted exercise with personalized dietary–nutritional therapy is the ideal approach for treating uremic sarcopenia. The key interventions for preventing and treating sarcopenia in CKD include aerobic and resistance exercises, along with personalized dietary–nutritional interventions. However, the effectiveness of these interventions in treating sarcopenia and preventing clinical consequences in CKD patients is still being determined
[72][74][180,182].