Energy is needed by cancer cells to stay alive and communicate with their surroundings. The primary organelles for cellular metabolism and energy synthesis are mitochondria. Researchers recently proved that cancer cells can steal immune cells’ mitochondria using nanoscale tubes. This finding demonstrates the dependence of cancer cells on normal cells for their living and function. It also denotes the importance of mitochondria in cancer cells’ biology. Emerging evidence has demonstrated how mitochondria are essential for cancer cells to survive in the harsh tumor microenvironments, evade the immune system, obtain more aggressive features, and resist treatments. For instance, functional mitochondria can improve cancer resistance against radiotherapy by scavenging the released reactive oxygen species. Therefore, targeting mitochondria can potentially enhance oncological outcomes, according to this notion. The tumors’ responses to anticancer treatments vary, ranging from a complete response to even cancer progression during treatment. Therefore, personalized cancer treatment is of crucial importance. So far, personalized cancer treatment has been based on genomic analysis. Evidence shows that tumors with high mitochondrial content are more resistant to treatment. This paper illustrates how mitochondrial metabolism can participate in cancer resistance to chemotherapy, immunotherapy, and radiotherapy. Pretreatment evaluation of mitochondrial metabolism can provide additional information to genomic analysis and can help to improve personalized oncological treatments. This article outlines the importance of mitochondrial metabolism in cancer biology and personalized treatments.
Energy is needed by cancer cells to stay alive and communicate with their surroundings. The primary organelles for cellular metabolism and energy synthesis are mitochondria. Researchers recently proved that cancer cells can steal immune cells’ mitochondria using nanoscale tubes. This finding demonstrates the dependence of cancer cells on normal cells for their living and function. It also denotes the importance of mitochondria in cancer cells’ biology. Emerging evidence has demonstrated how mitochondria are essential for cancer cells to survive in the harsh tumor microenvironments, evade the immune system, obtain more aggressive features, and resist treatments. For instance, functional mitochondria can improve cancer resistance against radiotherapy by scavenging the released reactive oxygen species. Therefore, targeting mitochondria can potentially enhance oncological outcomes, according to this notion. The tumors’ responses to anticancer treatments vary, ranging from a complete response to even cancer progression during treatment. Therefore, personalized cancer treatment is of crucial importance. So far, personalized cancer treatment has been based on genomic analysis. Evidence shows that tumors with high mitochondrial content are more resistant to treatment.
- mitochondria
- personalized oncology
- cancer stem cell
1. Introduction
2. The Pivotal Role of Mitochondria in Cancer Cells’ Metabolism
Cancer cells rely on functional mitochondria to survive in the harsh tumor microenvironment (TME), evade the immune system, progress to less differentiated types, and resist different treatment modalities [10], as follows: (Figure 1)- (A)
-
Surviving in the TME via the following mechanism:
-
- (A1)
-
Metabolic switch to glycolysis: cancer cells are reorganized to tolerate the hypoxic, acidic, and hypoglycemic conditions of TME. Hypoxia-inducible factor-1α (HIF-1α) is one of the primary regulators of this metabolic alteration. In the harsh TME, HIF-1α overexpression leads to a metabolic switch from oxidative phosphorylation (OxPhos) into glycolysis. This alteration can maintain the cellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) level in the hypoxic TME. It has been demonstrated that HIF-1α relies on functional mitochondria for a secure continuous function [11]. In 2020, van Gisbergen et al. realized that cancer cells with severe mitochondrial dysfunction showed a decrease in CAIX expression and HIF-1α levels. The authors concluded that functional mitochondria are essential for the stabilization of HIF-1α [11].
-
- (A2)
-
Scavenging reactive oxygen species (ROS): hypoxic condition of TME is associated with increased ROS production in cancer cells. When there is insufficient oxygen availability, the electron transport across the mitochondrial complexes is slowed down. This causes the electrons to leak out of the electron transport chain (ETC) and interact with oxygen, producing ROS. Functional mitochondria can detoxify the released ROS by preserving the cellular NADPH sources. This function is mediated by increased NADH production, representing mitochondrial function [12,13][12][13].
-
- (A3)
-
Arresting cell cycle: cancer cells can tolerate the harsh TME by dormancy, which is the mitotic arrest at the G0/G1 cycle phase [14]. Cell cycle progression is regulated by a dedicated system consisting of cyclins and cyclin-dependent kinases (CDK). It has been demonstrated that mitochondria can mediate dormancy in colon cancer cells by HIF-dependent activation of p21 and p27 (two CDK-cyclin inhibitors) [11[11][15],15], in prostate cancer cells by activating the MAPK-p38 pathway [16[16][17],17], and in leukemic stem cells by activating the mTOR pathway [18,19][18][19].
-
- (A4)
-
Maintaining pH homeostasis: In contrast to normal cells, cancer cells can tolerate acidic TME using a dedicated transmembrane glycoprotein called carbonic anhydrase IX (CA IX). This protein preserves intracellular pH by absorbing extracellular bicarbonate and sending out intracellular lactate [20,21][20][21]. It has been demonstrated that mitochondria are the upregulators of CA IX [11].
-
- (A5)
-
Mediating autophagy: mitochondria can facilitate autophagy by raising the level of intracellular ROS, which leads to the inactivation of mTORC1 (an autophagy inhibitor) and the activation of NRF2 (an autophagy activator) [22,23,24,25][22][23][24][25].
-
- (A6)
-
Angiogenesis: secretion of different angiogenic factors (e.g., VEGF, PGF, angiopoietin) in cancer cells is HIF-dependent [26]. Mitochondria conduct angiogenesis by securing HIF function [11].
-
- (A7)
-
Mitochondrial hijacking: cancer cells can steal mitochondria from T cells (and NK cells) via nano-scale tubes. Saha et al. demonstrated that this process is GTP-dependent [2]. Functional mitochondria can secure mitochondria hijacking by providing GTP from their TCA cycle [27].
-
- (B)
-
Immune evasion: completed via facilitating TME acidification, glucose influx, PD-1 upregulation on T cells (by mitochondrial hijacking) [28], recruiting myeloid-derived suppressor cells (MDSCs), PD-L1 overexpression on cancer cells (via STING-IFN pathway), MHC-1 downregulation, and the secretion of immunosuppressants [10]. Additionally, T cells’ mitochondrial hijacking leads to PD-1 upregulation on T-cells and depletes their energy to provide long-term cancer-fighting action [28].
- (C)
-
Aggressiveness: mitochondria are crucial for cancer progression via mediating genomic instability, quiescence evasion, and epithelial-to-mesenchymal transition (EMT) [10]. An increase in cellular ROS is the most common promoter of these three processes. Genomic instability is mediated by an increase in ROS levels and damage to nuclear nucleosides and inducing minority MOMP (mitochondrial outer membrane permeabilization) [10]; quiescence evasion is conducted by an increase in cellular ROS and following the activation of the Ras pathway [29,30][29][30]. ROS is a double-edged sword, destroying cancer cells at high levels and promoting cancer progression at moderate levels. Functional mitochondria help cancer cells to maintain cellular ROS at higher levels (so-called “elevated ROS balance”), facilitating cancer progression without damage to the cellular structures [31].
- (D)
-
Treatment resistance: mitochondria can protect cancer cells from chemotherapy and RT by eliminating the released ROS. Additionally, they increase chemotherapy resistance by encouraging the function of efflux pumps (by providing ATP) and inducing cell cycle arrest. Additionally, mitochondrial hijacking from T cells impairs the long-term effects of anti-PD-1 treatment [10].
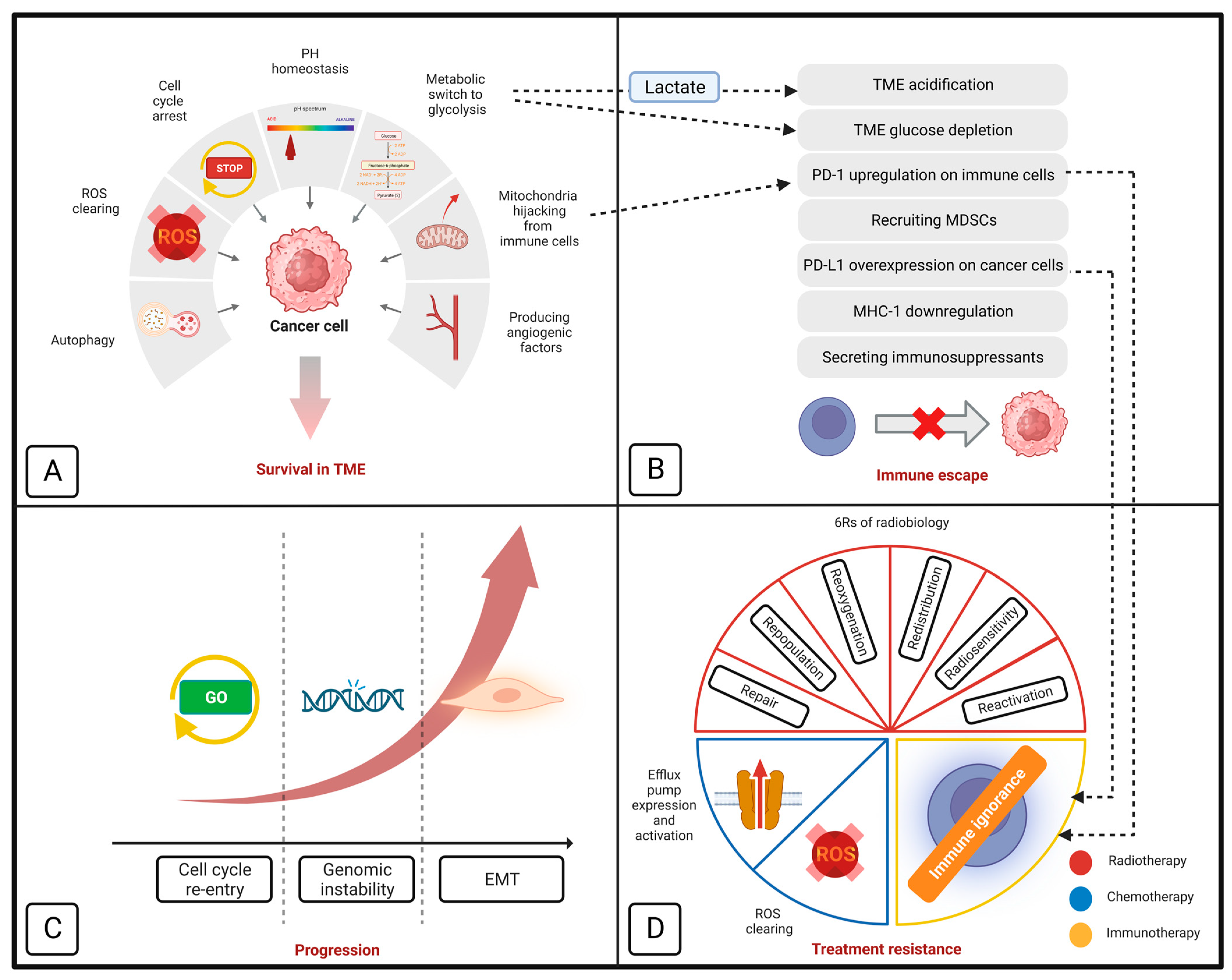 Figure 1. Schematic model of how mitochondria contribute to cancer cells’ survival in tumor microenvironment (A), immune evasion (B), progression (C), and resistance to different treatment modalities (D). Section D also demonstrates the importance of mitochondrial metabolism in ‘6Rs’ of radiobiology. EMT indicates epithelial-mesenchymal transition; MDSC, myeloid-derived suppressor cell; MHC-1, major histocompatibility complex class I; PD-1, programmed cell death protein-1; PD-L1, programmed cell death protein-ligand 1; ROS, reactive oxygen species; and TME, tumor microenvironment.
Figure 1. Schematic model of how mitochondria contribute to cancer cells’ survival in tumor microenvironment (A), immune evasion (B), progression (C), and resistance to different treatment modalities (D). Section D also demonstrates the importance of mitochondrial metabolism in ‘6Rs’ of radiobiology. EMT indicates epithelial-mesenchymal transition; MDSC, myeloid-derived suppressor cell; MHC-1, major histocompatibility complex class I; PD-1, programmed cell death protein-1; PD-L1, programmed cell death protein-ligand 1; ROS, reactive oxygen species; and TME, tumor microenvironment.3. Mitochondria Individualized Role in Cancer Metastasis
Metastasis happens in a very diverse and individualized pattern [33][32]. The players in the molecular pathway of metastasis and the therapeutic response to metastasis should also be considered in a personalized and idealized context. In order for cancer cells to spread, they must first undergo EMT, during which they lose intercellular adhesions and obtain high capacity for local migration, vascular invasion, and resistance to apoptotic stimuli. [34][33]. It has been found that there is a link between EMT and the stemness of cancer cells. These two processes are controlled by common mediators such as HIFs, SNAIL, and SLUG/SOX9 [35,36][34][35]. More functional mitochondria can promote EMT through releasing more mitochondrial ROS (mtROS), which activates different pathways, such as MAPK PI3K/Akt/mTOR, and VEGFA–SOX2–SNAI2 pathways [36,37,38][35][36][37]. Moreover, it is essential to acknowledge that mitochondria are directly involved in the cancer cells’ proliferation, invasion, and metastasis by enabling the linkage between β1 integrin and the extracellular matrix [39][38]. This process is mediated by lysyl oxidase (LOX), which requires HIF-1α for a secured function. Mitochondria can promote this process by promoting HIF-1α stability [11,40][11][39]. It is of utmost importance to employ targeted anti-mitochondrial to impede the process of EMT and curb the spread of cancer cells throughout the body. This approach can prove to be instrumental in arresting the progression of cancer and enhancing the effectiveness of treatment. Precisional targeting of cancer-specific mitochondria can reduce their ability to de-differentiate, proliferate, and metastasize, and helps to improve the treatment results and overall prognosis.4. Targeting Mitochondria: A Practical Strategy for Personalized Cancer Treatment
Thanks to the developments in medical genetics and molecular biology, the function of mitochondria in several cellular functions, including apoptosis, redox balance, macromolecule production, and calcium homeostasis, has been demonstrated [41,42][40][41]. In contrast to the ancient Warburg theory, the mitochondria of cancer cells are functional, supporting their survival and function [10]. As noted earlier, mitochondria can contribute to the development, progression, and metastasis of cancer. In addition, it has a crucial role in treatment resistance. Functional mitochondria can help cancer cells to overcome chemotherapy effects by scavenging released ROS and activating multidrug resistance pumps [10]. Also, they can improve the resistance against immunotherapy, by inhibiting the immune cells’ entry to the TME by depleting the glucose content of TME, acidifying the TME, and mediating the mitochondria hijacking from immune cells [10,43][10][42]. Next, wresearchers outline how mitochondria can improve the cancer cells resistance against radiotherapy. In a recent study, Taghizadeh-Hesary et al. demonstrated that mitochondria have a contributing role in tumor response to radiotherapy. They demonstrated that mitochondria are involved in so-called 6Rs of radiobiology [32][43] (Figure 1). The details of this link were presented as follows: (a) Repair: DNA damage is the primary cause of RT’s cytotoxic effects. Cancer cells with improved DNA repair mechanisms can counteract this effect. Mitochondria can support ATP-dependent proteins responsible for DNA integrity-related, including PARP-1 [44], XRCC1 [45], ATM [46], and DNA ligases [47], by providing enough ATP molecules. (b) Repopulation: Mitochondria can support cancer cells proliferation by supplying the building materials, including nucleic acids, amino acids, and lipids through stabilizing HIF-1 and metabolic switching to glycolysis [48]. (c) Reoxygenation: HIF-1 can mediate tissue reoxygenation by promoting the expression of different angiogenic factors and shielding endothelial cells from radiation effects [49]. HIF-1 needs functional mitochondria to function properly [11]. Consequently, healthy mitochondria can aid in the reoxygenation of tumor tissue. (d) Redistribution: Cyclin-Cdk complexes carefully control the cancer cells’ cell cycle [50]. The radiosensitive phases of the cell cycle are G2 and M and the radioresistant phases are G1 and S [51]. Cell cycle progression depends on dynamic responses of mitochondria during the G1 and S phases, when mitochondria fuse to form a hyperactive network; after that, they undergo fission to ensure proper partitioning between the two daughter cells [50]. In addition, functional mitochondria can help the cell cycle progression by supplying enough energy [52]. (e) Reactivation: Cancer cells have the ability to avoid activated immune cells by using immune inhibitory molecules like programmed death-ligand 1 (PD-L1) [53]. It has been shown that in the hypoxic TME, HIF-1α mediates PD-L1 expression on cancer cells [54]. This process is supported by functioning mitochondria which help to stabilize HIF-1α [11]. On the other hand, Akbari et al. found a direct correlation between T cell mitochondrial capacity and the expression of PD-1. If T cells have limited mitochondrial capacity, they may experience an overexpression of PD-1, ultimately leading to inactivity [28]. The study conducted by Saha et al. conclusively showed that specific nanotubes enable cancer cells to hijack the mitochondria of T and NK cells [2]. Applying this strategy, cancer cells deliberately raise their PD-L1 levels while boosting PD-1 in immune cells to cunningly evade the immune system. (f) Radiosensitivity: Functional mitochondria can reduce the radiosensitivity of cancer cells by scavenging the released ROS and mediating the removal of damaged mitochondria, a process called mitophagy [32][43]. Hitherto, numerous biological factors have been linked to the intrinsic radiosensitivity of cancer cells, including p53, transforming growth factor beta (TGF-β), and isocitrate dehydrogenase 1 (IDH1) among others. For instance, p53 can improve radioresistance by enhancing the mitochondrial DNA integrity and PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α) overexpression [55,56][55][56]. For the detailed mechanisms of other corresponding factors, the readers are referred to the study by Taghizadeh-Hesary et al. [32][43] (Table 1).Note: This Table is retrieved from the Taghizadeh-Hesary et al. study [32][43]. Abbreviations: 8-oxo-dG, 8-hydroxy-2′-deoxyguanosine; Akt, protein kinase B; AMPK, serine/threonine kinase AMP-activated protein kinase; ATM, ataxia-telangiectasia mutated; BRCA2, breast cancer gene 2; CAF, cancer-associated fibroblasts; FGFR1/3, fibroblast growth factor 1/3; HCC, hepatocellular carcinoma; HMGB1, high mobility group box 1; HOTAIR, HOX transcript antisense RNA; HR, homologous recombination; IDH1, Isocitrate dehydrogenase 1; KEAP1, Kelch-like ECH-associated protein; LAPTM4B, lysosome-associated transmembrane protein 4B; mt, mitochondrial; mTOR, mammalian target of rapamycin; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κB; NOTCH2, neurogenic locus notch homolog protein 2; NPC, nasopharyngeal carcinoma; NSCLC, non-small cell lung cancer; OxPhos, oxidative phosphorylation; PARP, poly (ADP-ribose) polymerase; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1α; PI3K, phosphoinositide 3-kinases; ROS, reactive oxygen species; RPA1, replication protein A1; RSK2, ribosomal S6 kinase; RT, radiotherapy; SCC, squamous cell carcinoma; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α; XRCC1, X-ray repair cross complementing 1.Table 1. The biological factors of radioresistance from the mitochondria perspective.Factors Cancer Ref. Interaction with Mitochondria Ref. Increasing radioresistance Mutated P53 Various [57] - −
-
Mutated p53 preserves mtDNA integrity
- −
-
Mutated p53 improves mt capacity (PGC1α-mediated)
- −
-
More functional mt scavenge more RT-induced ROS
[55]
[56]
[10]TGF-β HCC [58] - −
-
TGF-β signaling in CAFs mediates reverse Warburg effect
- −
-
CAFs’ lactate and pyruvate feed cancer cells’ mt OxPhos
- −
-
Activated OxPhos helps to restore NADPH
- −
-
NADPH supports the antioxidant defense system
[59]
[60]
[61]
[62]IDH1 Glioblastoma [63] - −
-
Mutated IDH1 enhances mt OxPhos (ROS generation)
- −
-
Mutated IDH1 downregulates cytochrome c
- −
-
Cytochrome c can nullify ROS
- −
-
Thus, IDH1 mutation disrupts the ROS balance
[64]
[65]
[66]PARP Breast
Ovarian
Prostate
Pancreatic
HCC[67]
[68]- −
-
PARP requires RAD51 for HR
- −
-
BRCA2 regulates RAD51 function
- −
-
BRCA2 requires mt support
- −
-
Thus, functional mt improves radioresistance by mediating HR
[69]
[69]
[70]PI3K/Akt/mTOR pathway Prostate [71] - −
-
mTOR upregulates mt proteins responsible for mt metabolism
- −
-
More functional mt scavenge more RT-induced ROS
[72]
[10]Wnt/β-catenin pathway Esophageal SCC [73] - −
-
Wnt upregulates HMGB1
- −
-
HMGB1 activates mitochondria
- −
-
More functional mt scavenge more RT-induced ROS
[73]
[74]
[10]NF-κB pathway Breast
Glioma
HCC
Melanoma
NSCLC[75] - −
-
Enhances mt respiration
- −
-
Regulates mt dynamics
- −
-
Regulates mt gene expression
[76] 8-oxo-dG Esophageal
Gastric[77] - −
-
Serum 8-oxo-DG level represents cellular ROS
- −
-
Cellular ROS is dependent on mt metabolism
[77]
[10]ATM Glioma [78] - −
-
Preserves mtDNA
[79] XRCC1 NSCLC
HNC[80] - −
-
Preserves mt respiratory chain
[81] NOTCH2 NSCLC [82] - −
-
Regulates mitochondrial function
[83] KEAP1 NSCLC [82] - −
-
Regulates mitochondrial function
- −
-
Regulates mitophagy
[84]
[85]FGFR1/3 NSCLC [82] - −
-
Regulates mitochondrial energy metabolism
[86] HOTAIR Breast [87] - −
-
Regulates mitochondrial function
[88]
[89]AMPK Glioblastoma [90] - −
-
Preserves mt biogenesis upon energy stress
[91] RPA1 Glioblastoma [92] - −
-
Preserves mtDNA
[93] RSK2 NSCLC [94] - −
-
Stimulates mt OxPhos
[95] LAPTM4B NPC [96] - −
-
Activates mTOR
- −
-
mTOR upregulates mt proteins responsible for mt metabolism
- −
-
More functional mt scavenge more RT-induced ROS
[97]
[72]
[10]Decreasing radioresistance TNFα NSCLC [98] - −
-
Impairs mt complex I and III
- −
-
Complex III is essential for NADPH activity
- −
-
Thus, reduces mt capacity to scavenge RT-induced ROS
[99]
[100]
-
-
-
-
-
-
-
-
References
- Trapani, D.; Franzoi, M.A.; Burstein, H.J.; Carey, L.A.; Delaloge, S.; Harbeck, N.; Hayes, D.F.; Kalinsky, K.; Pusztai, L.; Regan, M.M.; et al. Risk-adapted modulation through de-intensification of cancer treatments: An ESMO classification. Ann. Oncol. 2022, 33, 702–712.
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106.
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693.
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137.
- Overgaard, J.; Aznar, M.C.; Bacchus, C.; Coppes, R.P.; Deutsch, E.; Georg, D.; Haustermans, K.; Hoskin, P.; Krause, M.; Lartigau, E.F.; et al. Personalised radiation therapy taking both the tumour and patient into consideration. Radiother. Oncol. 2022, 166, A1–A5.
- Abdelkarem, O.A.I.; Choudhury, A.; Burnet, N.G.; Summersgill, H.R.; West, C.M.L. Effect of Race and Ethnicity on Risk of Radiotherapy Toxicity and Implications for Radiogenomics. Clin. Oncol. 2022, 34, 653–669.
- Ameri, A.; Heydarirad, G.; Choopani, R.; Poshtmahi, S.; Ameri, P.; Talebi, F.; Bagheri Pour, A.; Taghizadeh-Hesary, F. Sumac-rose water mouthwash versus benzydamine to prevent radiation-induced oral mucositis in head and neck cancers: A phase II randomized trial. J. Cancer Res. Clin. Oncol. 2023, 149, 7427–7439.
- Ameri, A.; Rahnama, N.; Talebi, F.; Sourati, A.; Taghizadeh-Hesary, F. An evaluation of cancer aging research group (CARG) score to predict chemotherapy toxicity in older Iranian patients with cancer. Oncologie 2023, 25, 223–232.
- Selvakumar, S.C.; Preethi, K.A.; Ross, K.; Tusubira, D.; Khan, M.W.A.; Mani, P.; Rao, T.N.; Sekar, D. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 2022, 21, 83.
- Taghizadeh-Hesary, F.; Akbari, H.; Bahadori, M.; Behnam, B. Targeted Anti-Mitochondrial Therapy: The Future of Oncology. Genes 2022, 13, 1728.
- van Gisbergen, M.W.; Offermans, K.; Voets, A.M.; Lieuwes, N.G.; Biemans, R.; Hoffmann, R.F.; Dubois, L.J.; Lambin, P. Mitochondrial Dysfunction Inhibits Hypoxia-Induced HIF-1α Stabilization and Expression of Its Downstream Targets. Front. Oncol. 2020, 10, 770.
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants 2021, 10, 1838.
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753.
- Druker, J.; Wilson, J.W.; Child, F.; Shakir, D.; Fasanya, T.; Rocha, S. Role of Hypoxia in the Control of the Cell Cycle. Int. J. Mol. Sci. 2021, 22, 4874.
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004, 23, 1949–1956.
- Mandal, J.P.; Shiue, C.N.; Chen, Y.C.; Lee, M.C.; Yang, H.H.; Chang, H.H.; Hu, C.T.; Liao, P.C.; Hui, L.C.; You, R.I.; et al. PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radic. Biol. Med. 2021, 163, 69–87.
- Yu-Lee, L.-Y.; Lee, Y.-C.; Pan, J.; Lin, S.-C.; Pan, T.; Yu, G.; Hawke, D.H.; Pan, B.-F.; Lin, S.-H. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci. Rep. 2019, 9, 18635.
- O’Reilly, E.; Zeinabad, H.A.; Szegezdi, E. Hematopoietic versus leukemic stem cell quiescence: Challenges and therapeutic opportunities. Blood Rev. 2021, 50, 100850.
- Shiau, J.P.; Chuang, Y.T.; Cheng, Y.B.; Tang, J.Y.; Hou, M.F.; Yen, C.Y.; Chang, H.W. Impacts of Oxidative Stress and PI3K/AKT/mTOR on Metabolism and the Future Direction of Investigating Fucoidan-Modulated Metabolism. Antioxidants 2022, 11, 911.
- Czowski, B.J.; Romero-Moreno, R.; Trull, K.J.; White, K.A. Cancer and pH Dynamics: Transcriptional Regulation, Proteostasis, and the Need for New Molecular Tools. Cancers 2020, 12, 2760.
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669.
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890.
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320.
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461.
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.W.; Gustafson, D.L.; Thorburn, A. Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition. Dev. Cell 2019, 50, 690–703.e696.
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839.
- Lambeth, D.O. What is the function of GTP produced in the Krebs citric acid cycle? IUBMB Life 2002, 54, 143–144.
- Akbari, H.; Taghizadeh-Hesary, F.; Bahadori, M. Mitochondria determine response to anti-programmed cell death protein-1 (anti-PD-1) immunotherapy: An evidence-based hypothesis. Mitochondrion 2022, 62, 151–158.
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67.
- Kadonosono, T.; Miyamoto, K.; Sakai, S.; Matsuo, Y.; Kitajima, S.; Wang, Q.; Endo, M.; Niibori, M.; Kuchimaru, T.; Soga, T.; et al. AGE/RAGE axis regulates reversible transition to quiescent states of ALK-rearranged NSCLC and pancreatic cancer cells in monolayer cultures. Sci. Rep. 2022, 12, 9886.
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Brunsell, T.H.; Langerud, J.; Berg, K.C.G.; Røsok, B.I.; Bjørnbeth, B.A.; Nesbakken, A.; Lothe, R.A.; et al. Metastatic heterogeneity of the consensus molecular subtypes of colorectal cancer. NPJ Genom. Med. 2021, 6, 59.
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412.
- Fazilaty, H.; Gardaneh, M.; Akbari, P.; Zekri, A.; Behnam, B. SLUG and SOX9 Cooperatively Regulate Tumor Initiating Niche Factors in Breast Cancer. Cancer Microenviron. 2016, 9, 71–74.
- Fazilaty, H.; Gardaneh, M.; Bahrami, T.; Salmaninejad, A.; Behnam, B. Crosstalk between breast cancer stem cells and metastatic niche: Emerging molecular metastasis pathway? Tumour Biol. 2013, 34, 2019–2030.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Kim, M.; Jang, K.; Miller, P.; Picon-Ruiz, M.; Yeasky, T.M.; El-Ashry, D.; Slingerland, J.M. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene 2017, 36, 5199–5211.
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226.
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729.
- Paupe, V.; Prudent, J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem. Biophys. Res. Commun. 2018, 500, 75–86.
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl. Oncol. 2021, 14, 100905.
- Houshyari, M.; Taghizadeh-Hesary, F. Is Mitochondrial Metabolism a New Predictive Biomarker for Antiprogrammed Cell Death Protein-1 Immunotherapy? JCO Oncol. Pract. 2022, 19, 123–124.
- Taghizadeh-Hesary, F.; Houshyari, M.; Farhadi, M. Mitochondrial metabolism: A predictive biomarker of radiotherapy efficacy and toxicity. J. Cancer Res. Clin. Oncol. 2023, 149, 6719–6741.
- Bentle, M.S.; Reinicke, K.E.; Bey, E.A.; Spitz, D.R.; Boothman, D.A. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J. Biol. Chem. 2006, 281, 33684–33696.
- Lévy, N.; Martz, A.; Bresson, A.; Spenlehauer, C.; de Murcia, G.; Ménissier-de Murcia, J. XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res. 2006, 34, 32–41.
- Kozlov, S.; Gueven, N.; Keating, K.; Ramsay, J.; Lavin, M.F. ATP activates ataxia-telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. J. Biol. Chem. 2003, 278, 9309–9317.
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338.
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757.
- Rakotomalala, A.; Escande, A.; Furlan, A.; Meignan, S.; Lartigau, E. Hypoxia in Solid Tumors: How Low Oxygenation Impacts the “Six Rs” of Radiotherapy. Front. Endocrinol. 2021, 12, 742215.
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165715.
- Syljuåsen, R.G. Cell cycle effects in radiation oncology. In Radiation Oncology; Wentz, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–8.
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol. 2017, 27, 69–81.
- Suwa, T.; Kobayashi, M.; Nam, J.-M.; Harada, H. Tumor microenvironment and radioresistance. Exp. Mol. Med. 2021, 53, 1029–1035.
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674.
- Ericson, N.G.; Kulawiec, M.; Vermulst, M.; Sheahan, K.; O’Sullivan, J.; Salk, J.J.; Bielas, J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012, 8, e1002689.
- Li, J.; Li, Y.; Chen, L.; Yu, B.; Xue, Y.; Guo, R.; Su, J.; Liu, Y.; Sun, L. p53/PGC-1α-mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol. Med. Rep. 2020, 22, 155–164.
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation. Oncol. Lett. 2021, 22, 661.
- Zheng, H.; Jarvis, I.W.H.; Bottai, M.; Dreij, K.; Stenius, U. TGF beta promotes repair of bulky DNA damage through increased ERCC1/XPF and ERCC1/XPA interaction. Carcinogenesis 2018, 40, 580–591.
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135.
- Hu, J.W.; Sun, P.; Zhang, D.X.; Xiong, W.J.; Mi, J. Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal 2014, 26, 2210–2216.
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 721–726.
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231.
- Tran, A.N.; Lai, A.; Li, S.; Pope, W.B.; Teixeira, S.; Harris, R.J.; Woodworth, D.C.; Nghiemphu, P.L.; Cloughesy, T.F.; Ellingson, B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol. 2014, 16, 414–420.
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 2021, 218, e20200924.
- Li, S.; Sun, C.; Gu, Y.; Gao, X.; Zhao, Y.; Yuan, Y.; Zhang, F.; Hu, P.; Liang, W.; Cao, K.; et al. Mutation of IDH1 aggravates the fatty acid-induced oxidative stress in HCT116 cells by affecting the mitochondrial respiratory chain. Mol. Med. Rep. 2019, 19, 2509–2518.
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 2000, 275, 37159–37166.
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601.
- Guillot, C.; Favaudon, V.; Herceg, Z.; Sagne, C.; Sauvaigo, S.; Merle, P.; Hall, J.; Chemin, I. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitrohepatocellular carcinoma models. BMC Cancer 2014, 14, 603.
- Singh, D.D.; Parveen, A.; Yadav, D.K. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines 2021, 9, 1512.
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82.
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437.
- De la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373.
- Zhao, Y.; Yi, J.; Tao, L.; Huang, G.; Chu, X.; Song, H.; Chen, L. Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 9, 433.
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; Van Houten, B.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711.
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340.
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154.
- Pour Khavari, A.; Liu, Y.; He, E.; Skog, S.; Haghdoost, S. Serum 8-Oxo-dG as a Predictor of Sensitivity and Outcome of Radiotherapy and Chemotherapy of Upper Gastrointestinal Tumours. Oxid. Med. Cell Longev. 2018, 2018, 4153574.
- Zhou, W.; Sun, M.; Li, G.H.; Wu, Y.Z.; Wang, Y.; Jin, F.; Zhang, Y.Y.; Yang, L.; Wang, D.L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol. Rep. 2013, 30, 1793–1801.
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Investig. 2007, 117, 2723–2734.
- Vaezi, A.; Feldman, C.H.; Niedernhofer, L.J. ERCC1 and XRCC1 as biomarkers for lung and head and neck cancer. Pharmgenomics Pers. Med. 2011, 4, 47–63.
- Xu, C.; Xu, J.; Ji, G.; Liu, Q.; Shao, W.; Chen, Y.; Gu, J.; Weng, Z.; Zhang, X.; Wang, Y. Deficiency of X-ray repair cross-complementing group 1 in primordial germ cells contributes to male infertility. FASEB J. 2019, 33, 7427–7436.
- He, K.; Zhang, S.; Pang, J.; Yin, J.; Zhang, J.; Mu, D.; Tang, S.; Li, L.; Bao, H.; Wu, X. Genomic Profiling Reveals Novel Predictive Biomarkers for Chemo-Radiotherapy Toxicity and Efficacy in Non-Small-Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, e437.
- Fan, F.; Zhuang, J.; Zhou, P.; Liu, X.; Luo, Y. MicroRNA-34a promotes mitochondrial dysfunction-induced apoptosis in human lens epithelial cells by targeting Notch2. Oncotarget 2017, 8, 110209–110220.
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97.
- Zeb, A.; Choubey, V.; Gupta, R.; Kuum, M.; Safiulina, D.; Vaarmann, A.; Gogichaishvili, N.; Liiv, M.; Ilves, I.; Tämm, K.; et al. A novel role of KEAP1/PGAM5 complex: ROS sensor for inducing mitophagy. Redox Biol. 2021, 48, 102186.
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine Phosphorylation of Mitochondrial Pyruvate Dehydrogenase Kinase 1 Is Important for Cancer Metabolism. Mol. Cell 2011, 44, 864–877.
- Zhang, S.; Wang, B.; Xiao, H.; Dong, J.; Li, Y.; Zhu, C.; Jin, Y.; Li, H.; Cui, M.; Fan, S. LncRNA HOTAIR enhances breast cancer radioresistance through facilitating HSPA1A expression via sequestering miR-449b-5p. Thorac. Cancer 2020, 11, 1801–1816.
- Zheng, P.; Xiong, Q.; Wu, Y.; Chen, Y.; Chen, Z.; Fleming, J.; Gao, D.; Bi, L.; Ge, F. Quantitative Proteomics Analysis Reveals Novel Insights into Mechanisms of Action of Long Noncoding RNA Hox Transcript Antisense Intergenic RNA (HOTAIR) in HeLa Cells. Mol. Cell Proteomics 2015, 14, 1447–1463.
- Kong, L.; Zhou, X.; Wu, Y.; Wang, Y.; Chen, L.; Li, P.; Liu, S.; Sun, S.; Ren, Y.; Mei, M. Targeting HOTAIR induces mitochondria related apoptosis and inhibits tumor growth in head and neck squamous cell carcinoma in vitro and in vivo. Curr. Mol. Med. 2015, 15, 952–960.
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem. Biophys. Res. Commun. 2022, 590, 82–88.
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135.
- Pedersen, H.; Anne Adanma Obara, E.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1588.
- Liu, X.; Shan, G. Mitochondria Encoded Non-coding RNAs in Cell Physiology. Front. Cell Dev. Biol. 2021, 9, 713729.
- Ma, J.; Lu, Y.; Zhang, S.; Li, Y.; Huang, J.; Yin, Z.; Ren, J.; Huang, K.; Liu, L.; Yang, K.; et al. β-Trcp ubiquitin ligase and RSK2 kinase-mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ. 2018, 25, 1473–1485.
- Mrozowski, R.M. Targeting the Ser/Thr Protein Kinase RSK to Reduce Breast Cancer Metastasis; Vanderbilt University: Nashville, TN, USA, 2015.
- Chu, C.; Niu, X.; Ou, X.; Hu, C. LAPTM4B knockdown increases the radiosensitivity of EGFR-overexpressing radioresistant nasopharyngeal cancer cells by inhibiting autophagy. Onco Targets Ther. 2019, 12, 5661–5677.
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250.
- Pal, S.; Yadav, P.; Sainis, K.B.; Shankar, B.S. TNF-α and IGF-1 differentially modulate ionizing radiation responses of lung cancer cell lines. Cytokine 2018, 101, 89–98.
- Shinde, A.; Jung, H.; Lee, H.; Singh, K.; Roy, M.; Gohel, D.; Kim, H.B.; Mane, M.; Vasiyani, H.; Currim, F.; et al. TNF-α differentially modulates subunit levels of respiratory electron transport complexes of ER/PR +ve/−ve breast cancer cells to regulate mitochondrial complex activity and tumorigenic potential. Cancer Metab. 2021, 9, 19.
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181.