Daylight photodynamic therapy (dPDT) uses sunlight as a light source to treat superficial skin cancer. Using sunlight as a therapeutic device has been present for centuries, forming the basis of photodynamic therapy in the 20th century. Compared to conventional PDT, dPDT can be a less painful, more convenient and an effective alternative. The first clinical uses of dPDT on skin cancers began in Copenhagen in 2008. Currently, aminolevulinic acid-mediated dPDT has been approved to treat actinic keratosis patients in Europe.
- photodynamic therapy
- actinic keratosis
- dermatology
- skin cancer
Please note: Below is an entry draft based on your previous paper, which is wrirren tightly around the entry title. Since it may not be very comprehensive, we kindly invite you to modify it (both title and content can be replaced) according to your extensive expertise. We believe this entry would be beneficial to generate more views for your work. In addition, no worry about the entry format, we will correct it and add references after the entry is online (you can also send a word file to us, and we will help you with submitting).
Definition
Daylight photodynamic therapy (dPDT) uses sunlight as a light source to treat superficial skin cancer. Using sunlight as a therapeutic device has been present for centuries, forming the basis of photodynamic therapy in the 20th century. Compared to conventional PDT, dPDT can be a less painful, more convenient and an effective alternative. The first clinical uses of dPDT on skin cancers began in Copenhagen in 2008. Currently, aminolevulinic acid-mediated dPDT has been approved to treat actinic keratosis patients in Europe.
1. Introduction
Photodynamic therapy (PDT) is a medical treatment utilizing photosensitizers in conjunction with a specific light source to exert cytotoxic activity in tumor cells [1]. PDT has been approved to treat superficial nonmelanoma skin cancer worldwide, to date, due to its noninvasive procedure, enhanced tumor selectivity, good to excellent cosmetic outcome and large treatment field [2,3][2][3]. Photosensitizers (PS), visible light and oxygen are the three key elements in PDT, and the combination of the three results in tumor necrosis and apoptosis. PS is excited by specific wavelengths that contain the absorption peaks of a PS in visible light (usually red or blue light), near-infrared light and even sunlight. After illumination, the PS is excited from the ground state to the triplet state (Figure 1). The excited photosensitizer (PS*) undergoes two kinds of reactions. In type I reaction, the excited PS reacts with biomolecules such as lipids, proteins and amino acids to yield the superoxide anion radical (O2•−) and HO2•, through electron transfer. O2•− undergoes dismutation to form hydrogen peroxide (H2O2), the precursor of the highly reactive hydroxyl radical (OH•). OH• is extremely chemically reactive to almost all biological molecules, which can achieve a better antihypoxia outcome [4]. In the type II reaction, the excited PS yields singlet oxygen (1O2) through direct energy transfer to molecular oxygen. Singlet oxygen, like the hydroxyl radical, is highly reactive. The two reactions may occur simultaneously, and the ratio of the reactions depends on the type of PS used and concentrations of substrate and oxygen. Nonetheless, the type II reaction is the principal mechanism of O2-dependent PDT (Figure 1) [5,6,7][5][6][7]. PDT with PS, especially 5-aminolaevulinic acid (5-ALA) or its ester form, methyl aminolevulinate (MAL), is widely applied to dermatologic diseases including superficial nonmelanoma skin cancers, infections, infestation diseases, inflammatory diseases and photorejuvenation. The major limitation of PDT in treating skin diseases, either benign or malignant, is the penetration depth of light. Figure 2 shows the relationship between wavelengths of light and skin penetration. As a result, PDT is approved to treat actinic keratosis (AK) and nonmelanoma skin cancers including Bowen’'s disease (squamous cell carcinoma in situ), superficial basal cell carcinomas and certain thin basal cell carcinomas. Other emerging indications include extramammary Paget’'s disease, cutaneous Leishmaniasis, verruca, acne vulgaris, scleroderma and vulvar lichen sclerosis et atrophicus [3].
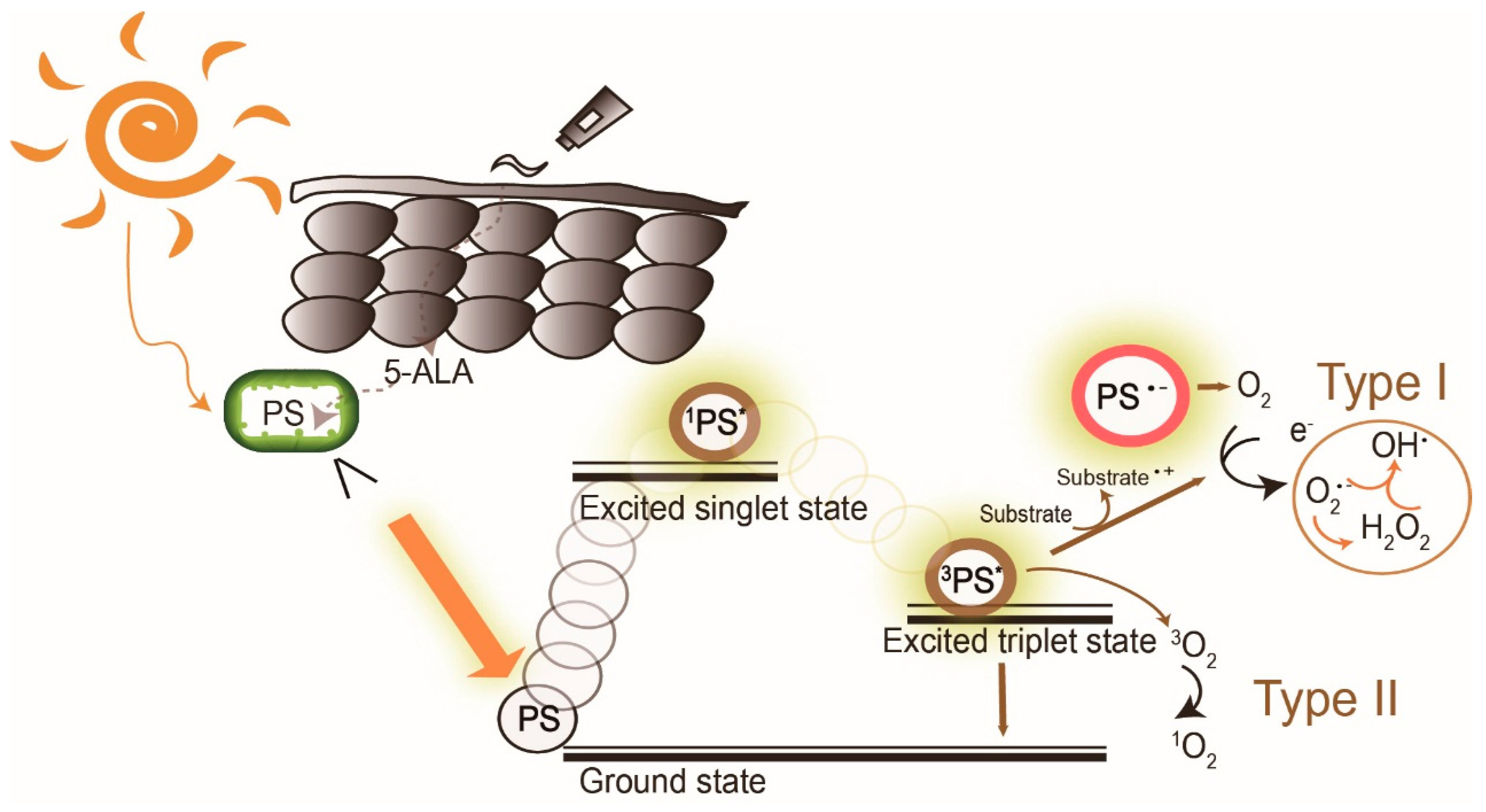
The mechanism of daylight photodynamic therapy. After daylight absorption, the photosensitizer (PS, 5-aminolevulinic acid (5-ALA), a prodrug of the real PS protoporphyrin IX, is exemplified here) is excited to a singlet state and undergoes intersystem crossing to the excited triplet-state. The triplet excited PS can react in two ways: a Type I reaction which involves the generation of superoxide anion radical (O
), hydrogen peroxide (H
O
), and hydroxyl radical (OH
) by electron transfer to molecular oxygen, and/or by the type II reaction through energy transfer to generate singlet oxygen (
O2). PS: photosensitizer;
PS*: excited singlet state,
PS* excited triplet state.
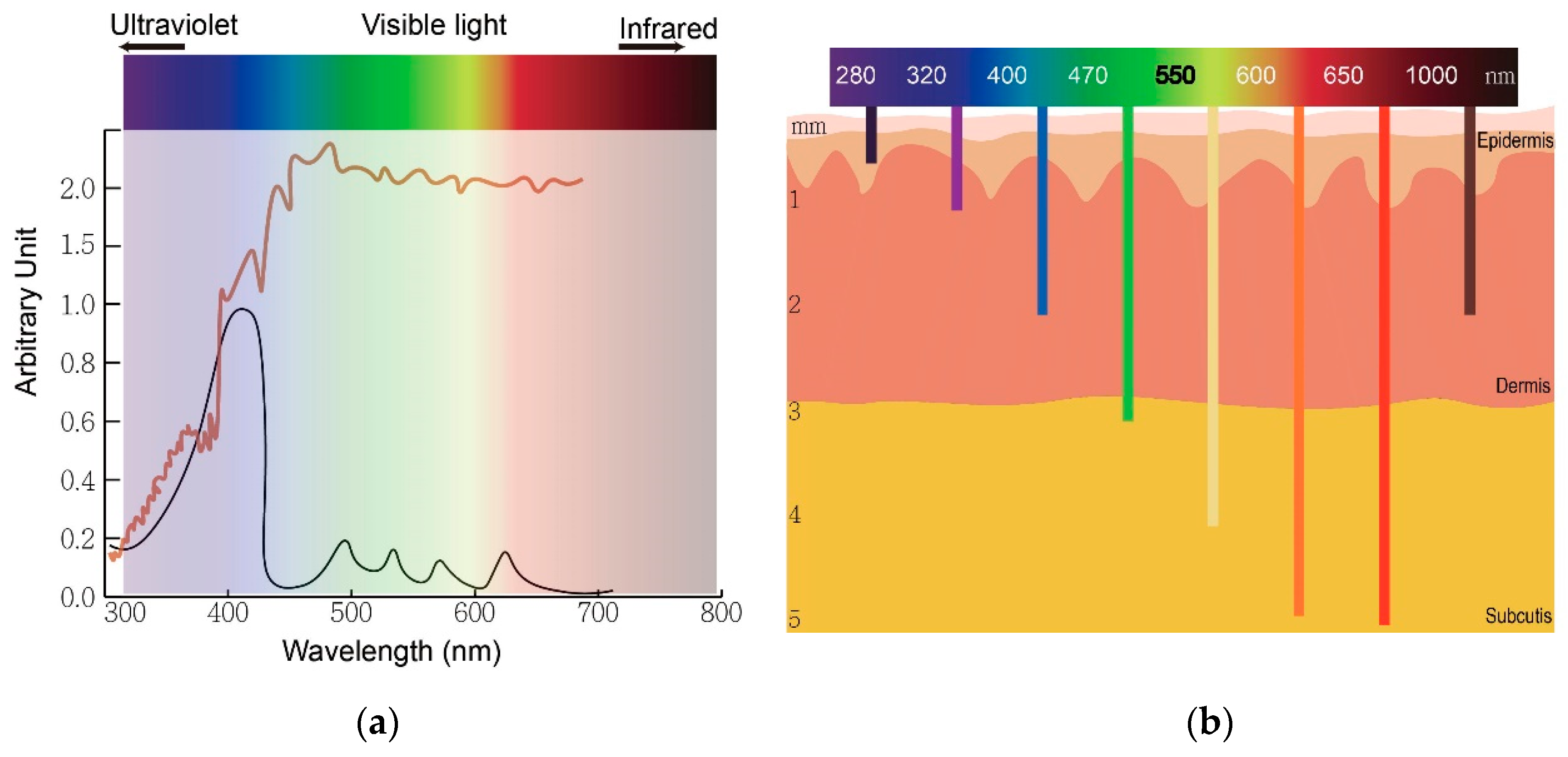
The wavelength of light determines an optimal therapeutic window of photodynamic therapy. (
) The absorption peaks of protoporphyrin IX (black) and sunlight spectrum (brown). (
) The relationship between wavelengths of light and skin penetration depth.
Actinic keratosis (AK) is a common precancerous skin lesion caused by cell damage under chronic exposure to ultraviolet (UV) light from sunlight or indoor tanning. People with fair skin color (Fitzpatrick skin types I and II) are more susceptible to AK. In Australia, the prevalence of AK was estimated to be 40–50% of the population 40 years and older [8]. The dysplastic keratinocytes in AK are confined to the basal layer of the epidermis in the skin, usually less than 300 μm deep. This depth is easily reached by light with wavelengths longer than 400 nm (blue) and renders AK the first skin disease approved for PDT treatment globally. Clinically, AK lesions are dry, scaly and erythematous spots or patches located on sun-damaged skin. The AK lesions are often more easily felt than seen. They are rough and dry to touch. PDT allows treatment of large and multiple lesions. The treatment outcome is not inferior to conventional therapies such as surgery and cryotherapy while the cosmetic outcome is superior to these conventional treatments.
In conventional PDT (cPDT), after the application of prodrug ALA for 3 h, the metabolic product protoporphyrin (PpIX) accumulates in the mitochondria of a cell. PpIX is photoactivated by irradiation with specific wavelengths of light. The light source must correspond with the absorption peaks of PpIX. PpIX has absorption peaks at 405, 510, 545, 580, 635 nm (Figure 2a). The Soret band of PpIX is 405 nm but a 635 nm light source is widely used in clinics for the purpose of a deeper light penetration. Blue (410 nm) and red (635 nm) light from lasers, lamps and LEDs are most commonly used. After irradiation, reactive oxygen species (ROS) by type I reaction and singlet oxygen by type II reaction cause cell death (Figure 1). The therapeutic depth from the skin is 1~2 mm with blue light and 1~6 mm with red light, limiting PDT usage in more superficial cutaneous lesions (Figure 2b). The light sources are not widely available in every clinic. cPDT is time-consuming (usually 3 h occlusion of ALA), staff skills dependent and relatively limited in the treatment area under a fixed light source. Although cPDT is a convenient field-directed therapy with a good cosmetic outcome, the discomfort and pain during and after cPDT impedes its usage. Daylight emits UV, visible and infrared lights which cover the absorption peaks of most, if not all, available photosensitizers nowadays (Figure 2b). Patients experienced less pain with daylight PDT (dPDT) and were more satisfied with the procedure. A 2-h daylight exposure is sufficient for treatment and weather does not have a large impact on dPDT in most countries.
2. History of PDT
The earliest known reports of phototherapy, previously known as heliotherapy, were in ancient Greece, Egypt and India in the treatment of various diseases such as vitiligo, psoriasis and skin cancer [9]. Hippocrates, the Father of Medicine, spread the teachings of heliotherapy, using the sun as a therapeutic agent. Phototherapy was popularized as a medical treatment by the Danish physician Niels Finsen who received the Nobel prize in 1903 for his work in carbon arc phototherapy for skin tuberculosis [10]. Incidental findings of toxicity on paramecium via fluorescence, a product of light and acridine together, were observed in an experiment led by Professor Herman von Tappeiner and his student, Oscar Raab. In 1904, Von Tappeiner and Joldbaur reported that oxygen was required for photosensitization. In 1907, Von Tappeiner first coined the term photodynamic therapy in his book narrating experiments of oxygen-dependent photosensitization on skin cancer, lupus of the skin and condyloma of the female genitalia [11].
Hematoporphyrin (hp) was first produced by Scherer in 1841 [12]. In 1908, Hausmann studied the phototoxic effects on white mice and found that the effects were dependent on the doses of photosensitizer and light. The first proof of porphyrins as photosensitizers applied on humans was in 1913 when Friedrich Meyer-Betz injected himself with 200 mg hematoporphyrin and experienced severe photosensitive skin reactions for two months [13]. In 1924, Policard first observed that UV radiation produced red fluorescence in experimental rat sarcomas and correctly attributed the finding to the accumulation of hp. In 1942, Auler and Banzer were the first to observe PDT with hp on tumors. An evolutionary finding was made by Schwartz, in 1995, who isolated hp derivatives (HpD) which were more enhanced in tumor-localizing properties and phototoxicity than hp itself. The selective and destructive effect of HpD on breast cancer and bladder tumors was investigated by Lipson et al. [14] and Kelly and Snell [15] in 1966 and 1976, respectively. The modern era of PDT began with the first systematic human trials for PDT on cutaneous and subcutaneous cancer performed by Dougherty et al. in 1978 [16]. Dougherty and his coworkers also purified HpD and produced Photofrin, which was the first clinically approved photosensitizer for the treatment of cancer.
cPDT with Photofrin was first approved by the US Food and Drug Administration (FDA) in 1995 [17]. Photofrin (Porfimer sodium; Axcan Pharma, Inc., Ontario, CA, USA) is injected intravenously and irradiated with 630 nm wavelength laser light [18]. However, increased skin photosensitivity persists over two months after the administration of Photofrin. Therefore, second-generation photosensitizers were developed over the decades. Currently, three ALA based photosensitizers are approved to treat skin cancers with PDT in the United States, including 5-ALA (Levulan®, DUSA Pharma Inc., New York, NY, USA), methyl-5 aminolevulinic acid (MAL) (Metvix®, Photocure ASA, Oslo, Norway) and a new nanoemulsion 10% ALA hydrogel (BF-200, Ameluz®, Biofrontera Bioscience GmbH, Overland Park, KS, USA). The second-generation photosensitizers are more effective and less harmful than Photofrin.
The original form of ALA in topical creams, ointments or gels is very unstable and needs to be used within hours to days after production due to its chemical properties [19]. ALA undergoes dimerization irreversibly to form 2,5-dicarboxyethyl-3,6-dihydropyrazine, which spontaneously oxidizes to 2,5-dicarboxyethylpyrazine in aqueous solutions. In addition, ALA is a zwitterion at physiological pH which impairs its penetration through the lipophilic stratum corneum [20]. From a pharmaceutical perspective, epidermal penetration and stability of the prodrug and its vehicle determine the efficacy of ALA-PDT [21]. By adding methyl esters, MAL became one of the more lipophilic ALA derivatives with better stability and deeper penetration than ALA. However, the ester is cleaved away before MAL can enter the heme biosynthetic pathway. As a result, less PpIX is formed with MAL as compared to ALA after the same incubation time [19]. A new 10% ALA hydrogel (BF-200) was developed [19]. The oil-in-water nanoemulsion hydrogel contains 7.8% of free ALA that connects to the polar phosphatidylcholine head groups of lipids and extends the ALA stability from several days to over two years [19]. The nanoemulsion BF-200 is approximately 20 nm, contains a monolayer of phosphatidylcholine molecules and a long chain of MiglyolR®812 with many caprylic acid (C8) and capric acid (C10) and fatty acid (FA) molecules. BF-200 nanovesicles rapidly fuse with stratum corneum (SC) lipid layers and the shorter FA integrates into the SC, increasing SC fluidity and ALA penetration through the epidermis [19]. Figure 3 demonstrates the evolution of ALA based photosensitizers. dPDT with MAL cream (Metvix®, TM, Photocure ASA, Oslo, Norway) and BF-200 ALA (Ameluz® Biofrontera, Leverkusen, Germany) are licensed to treat AK in Europe [22]. Dirschka et al. revealed a noninferiority of BF-200 ALA to MAL with dPDT in the treatment of mild-to-moderate AK [19,23][19][23]. At a three-month follow-up, 79.8 and 76.5% of the AK lesions treated with BF-200 ALA gel-dPDT and with MAL-dPDT showed complete remission, respectively. The BF-200 ALA gel group had lower recurrence rates after a one-year follow-up. A new self-adhesive 5-ALA patch form (Alacare®; Photonamic, Pinneberg, Germany) is approved to treat AK and is advantageous since pretreatment is unnecessary [3].
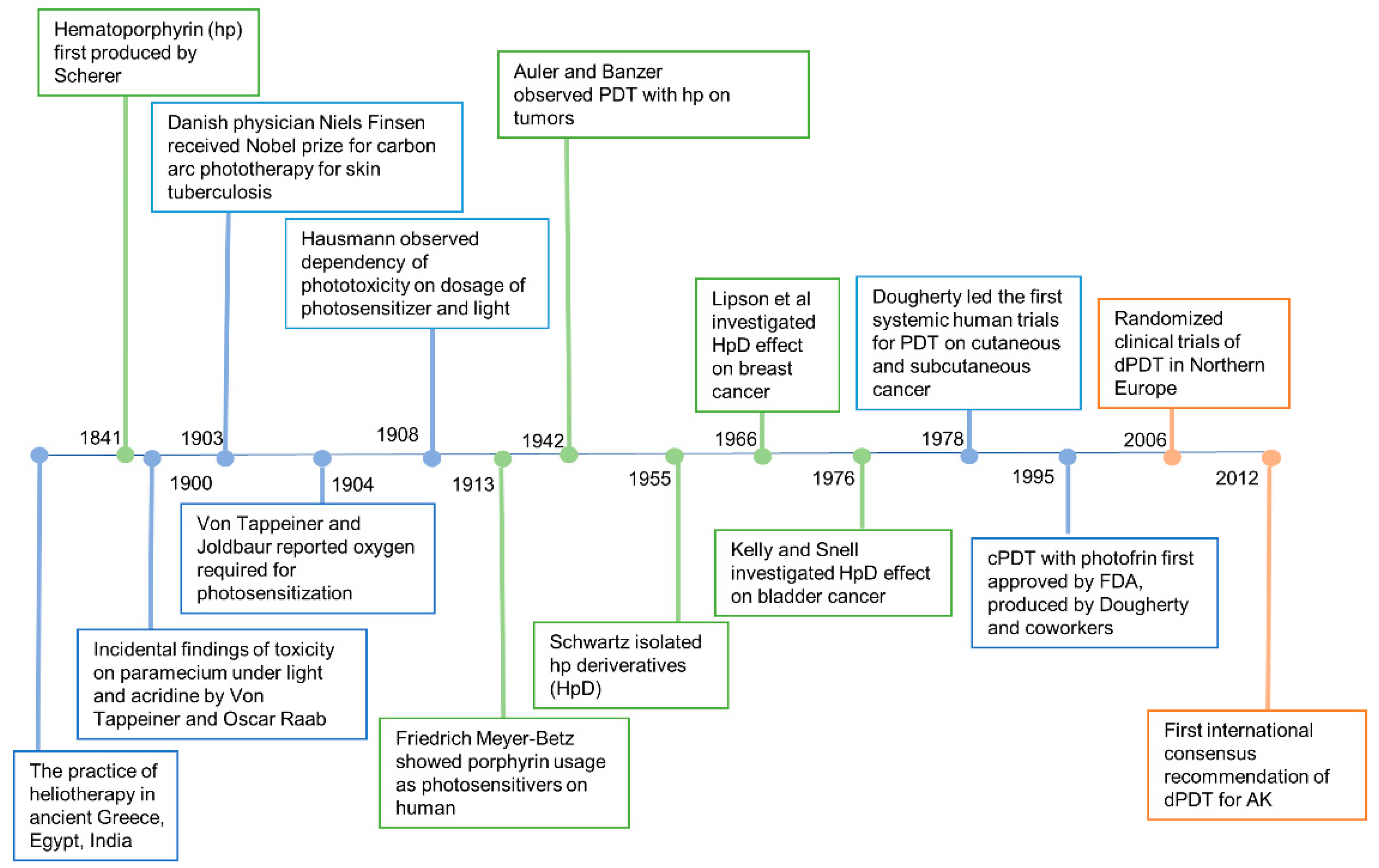
History of photodynamic therapy.
cPDT uses artificial light to activate the photosensitizers. However, due to the significant pain with cPDT, sunlight was considered an alternative light source. Weigel et al. led the pioneer work in northern Europe demonstrating the efficacy, safety and benefits of dPDT [24]. An international consensus recommendation of dPDT for actinic keratosis, penned by The International Society for Photodynamic Therapy in Dermatology, was published in 2012 [25]. Since then, consensus recommendations in Australia, UK, Spain and other countries have been made according to the geological and climatic changes in individual countries.
3. Selectivity of PDT on Cancer Cells
5-aminolevulinic acid (ALA) is the most common photosensitizer used in the dermatologic field nowadays. It is a prodrug that converts into protoporphyrin IX (PpIX) via an intrinsic cellular heme biosynthetic pathway (Figure 4). PpIX receives feedback inhibition by the intracellular level of heme. More exogenous PpIX accumulates in the inflammatory, premalignant and malignant cells than in normal epidermal cells due to the alterations in enzyme activity of heme biosynthesis. The limited capacity of ferrochelatase (FECH) and increased enzymatic activity of ALA dehydratase (ALAD), porphobilinogen deaminase (PBGD) and uroporphyrinogen decarboxylase (UROD) result in the accumulation of exogenous PpIX in tumor cells [26,27][26][27]. Another possible explanation for the increased accumulation in neoplastic cells could be altered ALA or porphyrin uptake or decreased PpIX efflux mechanisms. Recent studies have also suggested that enhanced glycolysis in tumor cells, and the subsequent switching from glucose tricarboxylic cycle acid (TCA) to aerobic glycolysis, may activate the heme biosynthesis pathway to remove the TCA metabolites [25]. The increased activation of the heme biosynthesis pathway may enhance PpIX accumulation due to limited FECH.
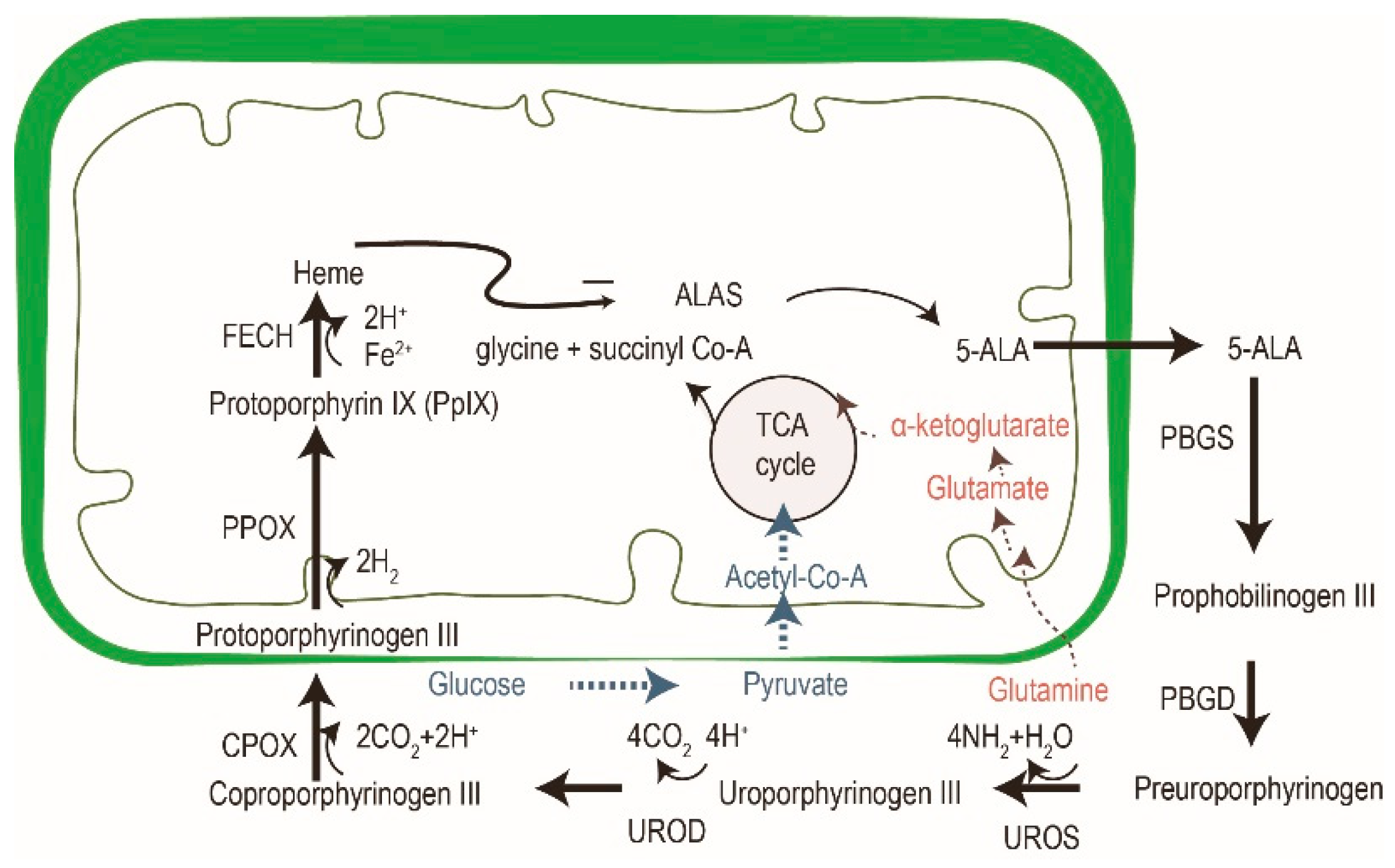
Figure 4. The cross-talk of glucose, glutamine and porphyrin heme biosynthesis pathways. In addition to porphyrin synthesis via the tricarboxylic cycle acid (TCA) cycle, the accumulation of PpIX in tumor cells might be explained by an increase of glycolysis (in blue) and glutaminolysis (in red). Modified from reference 24.
4. Advantages of dPDT
The major drawback of cPDT is the intolerable burning or stinging pain during or after treatment. Pain during irradiation was thought to be related to a large accumulation of PpIX after a long incubation time, especially on the acral and genital skin where nerve fibers are abundant [28,29,30][28][29][30]. The mechanism that causes pain with PDT is not well understood. Reactive oxygen species (ROS) were believed to be the main contributor in PDT-induced pain by increasing phosphorylation of N-methyl-d- involved in the pathway of nociception [31,32,33][31][32][33]. ROS-mediated tissue damage also results in the aspartate (NMDA), transient receptor potential vanilloid type 1 (TRPV1) and transient receptor potential ankyrin 1 (TRPA1) receptors release of proinflammatory cytokines including interleukin 1, 6 and tumor necrosis factor-alpha [31]. Both effects of ROS contribute to the stimulation of nociceptors. Others have postulated that local hypoxia, after depletion of oxygen in the process of PpIX photoactivation and tumor destruction, can decrease pH in tissue and trigger pain signals [33,34][33][34]. Many studies revealed more severe pain with ALA compared to MAL [35], perhaps since ALA is actively transported into the peripheral nerve endings, probably to the unmyelinated afferent c-fibers, and induces nerve stimulation during irradiation [26]. Whether pain increases with light fluences (J/cm2) or not is controversial [35]. Wang et al. theorized that the pain with PDT positively correlates with fluence rate (W/m2 or mW/cm2) and dose below a certain threshold (around 60 mW/cm2, 50 J/cm2) and reaches a plateau beyond the threshold, possibly due to the desensitization of nociceptors or limited cell capacity to produce ROS. Grapengiesser et al. found that patients who received cPDT on actinic keratosis (AK) lesions suffered more pain (higher visual analog scale (VAS)) than those treated on Bowen’'s disease and basal cell carcinoma (BCC) lesions [36]. Studies proposed that cPDT on well-innervated locations, such as the face, hands and perineal regions, were more painful [36]. Wiegell et al. proposed that continuous PpIX activation without occlusion and small accumulation of PpIX makes dPDT a less painful, but as effective, alternative to cPDT [24].
dPDT uses daylight as an alternative light source for PDT in the treatment of actinic keratosis (AK) and other skin lesions. dPDT emerged in Europe around 2006 [24]. In contrast with the three-hour incubation in cPDT, dPDPT only requires a 30-min incubation. The reason for the shortened incubation is the constant low-level activation of PpIX during daylight exposure. In cPDT, approximately 80% of PpIX is activated simultaneously within a few minutes to an illumination dose of 10 J/cm2 [37]. In dPDT, however, the continuous activation of PpIX matches the speed of PpIX formation. No PpIX is accumulated in the skin, contributing to the reduced pain during the treatment. The visible light (wavelength: 380 to 700 nm) is used in dPDT. The wavelength is different from UV light (100 to 380 nm,) thus sunscreen is recommended to block broadband UV to prevent UV damage under daylight exposure. dPDT is also more convenient as it can be performed by the patients themselves at home. An investigation conducted in the UK reported that patients were willing to perform dPDT at home with 82% being happy or very happy with the service due to minimal adverse events, good efficacy and cosmetic outcome [38]. Rubel et al. also revealed less scarring or hypopigmentation of dPDT compared to cPDT [39]. Not only is dPDT less painful and larger in the treatment area than cPDT but the incubation time is shorter than cPDT (Table 1).
Table 1. Advantages and disadvantages of cPDT versus dPDT.
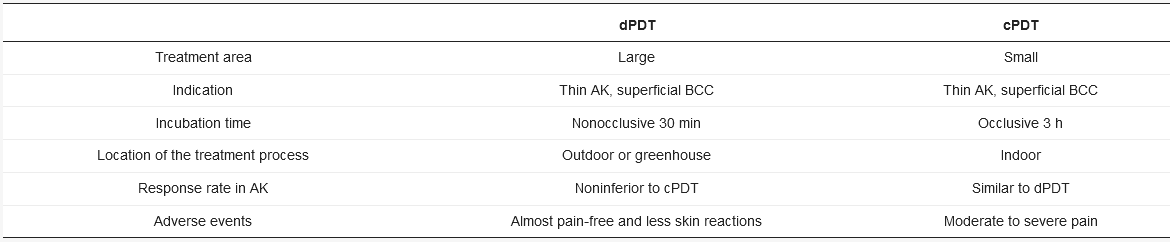
AK: actinic keratosis; BCC: basal cell carcinoma.
5. Conclusions
dPDT using natural sunlight to activate photosensitizers is advantageous in its lower cost and reduced pain. It is a promising method to treat AK under the concept of field cancerization. Current studies suggest that the prognosis of dPDT is noninferior to cPDT. dPDT is licensed to treat AK in Europe with the support of randomized trials and consensus guidelines. There are limited reports on PDT on other skin tumors or inflammatory skin diseases other than AK. Based on the history of the development of cPDT, we can expect that the application of dPDT will grow exponentially shortly. Further noninferiority studies between dPDT and cPDT are warranted.
References
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905, doi:10.1093/jnci/90.12.889.
- Lopez, R.F.; Lange, N.; Guy, R.; Bentley, M.V. Photodynamic therapy of skin cancer: Controlled drug delivery of 5-ALA and its esters. Adv. Drug Deliv. Rev. 2004, 56, 77–94, doi:10.1016/j.addr.2003.09.002.
- Morton, C.A.; Szeimies, R.M.; Basset-Seguin, N.; Calzavara-Pinton, P.; Gilaberte, Y.; Haedersdal, M.; Hofbauer, G.F.L.; Hunger, R.E.; Karrer, S.; Piaserico, S.; et al. European Dermatology Forum guidelines on topical photodynamic therapy 2019 Part 1: Treatment delivery and established indications—Actinic keratoses, Bowen’s disease and basal cell carcinomas. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 2225–2238, doi:10.1111/jdv.16017.
- Zhuang, Z.; Dai, J.; Yu, M.; Li, J.; Shen, P.; Hu, R.; Lou, X.; Zhao, Z.; Tang, B.Z. Type I photosensitizers based on phosphindole oxide for photodynamic therapy: Apoptosis and autophagy induced by endoplasmic reticulum stress. Chem. Sci. 2020, 11, 3405–3417, doi:10.1039/d0sc00785d.
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one-photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293, doi:10.1016/S1572-1000(05)00007-4.
- Noodt, B.B.; Berg, K.; Stokke, T.; Peng, Q.; Nesland, J.M. Apoptosis and necrosis induced with light and 5-aminolaevulinic acid-derived protoporphyrin IX. Br. J. Cancer 1996, 74, 22–29, doi:10.1038/bjc.1996.310.
- Ding, H.; Yu, H.; Dong, Y.; Tian, R.; Huang, G.; Boothman, D.A.; Sumer, B.D.; Gao, J. Photoactivation switch from type II to type I reactions by electron-rich micelles for improved photodynamic therapy of cancer cells under hypoxia. J. Control. Release 2011, 156, 276–280, doi:10.1016/j.jconrel.2011.08.019.
- Salasche, S.J. Epidemiology of actinic keratoses and squamous cell carcinoma. J. Am. Acad. Dermmatol. 2000, 42, S4–S7, doi:10.1067/mjd.2000.103342.
- Daniell, M.D.; Hill, J.S. A history of photodynamic therapy. Aust. N. Z. J. Surg. 1991, 61, 340–348, doi:10.1111/j.1445-2197.1991.tb00230.x.
- Urbach, F.; Forbes, P.D.; Davies, R.E.; Berger, D. Cutaneous photobiology: Past, present and future. J. Investig. Dermatol. 1976, 67, 209–224, doi:10.1111/1523-1747.ep12513042.
- Ackroyd, R.; Kelty, C.; Brown, N.; Reed, M. The history of photodetection and photodynamic therapy. Photochem. Photobiol. 2001, 74, 656–669.
- Ormond, A.B.; Freeman, H.S. Dye Sensitizers for Photodynamic Therapy. Materials 2013, 6, 817–840, doi:10.3390/ma6030817.
- Abdel-Kader, M.H. History of Photodynamic Therapy. In Photodynamic Therapy: From Theory to Application, Abdel-Kader, M.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–22, doi:10.1007/978-3-642-39629-8_1.
- Lipson, R.L.; Baldes, E.J.; Gray, M.J. Hematoporphyrin derivative for detection and management of cancer. Cancer 1967, 20, 2255–2257.
- Dougherty, T.J. Studies on the structure of porphyrins contained in Photofrin II. Photochem. Photobiol. 1987, 46, 569–573, doi:10.1111/j.1751-1097.1987.tb04815.x.
- Kessel, D. Photodynamic Therapy: A Brief History. J. Clin. Med. 2019, 8, doi:10.3390/jcm8101581.
- Huang, Z. A review of progress in clinical photodynamic therapy. Technol. Cancer Res. Treat. 2005, 4, 283–293, doi:10.1177/153303460500400308.
- Baskaran, R.; Lee, J.; Yang, S.G. Clinical development of photodynamic agents and therapeutic applications. Biomater. Res. 2018, 22, 25, doi:10.1186/s40824-018-0140-z.
- Reinhold, U. A review of BF-200 ALA for the photodynamic treatment of mild-to-moderate actinic keratosis. Future Oncol. 2017, 13, 2413–2428, doi:10.2217/fon-2017-0247.
- Tewari, K.M.; Eggleston, I.M. Chemical approaches for the enhancement of 5-aminolevulinic acid-based photodynamic therapy and photodiagnosis. Photochem. Photobiol. Sci. 2018, 17, 1553–1572, doi:10.1039/c8pp00362a.
- Tsai, J.C.; Chen, I.H.; Wong, T.W.; Lo, Y.L. In vitro/in vivo correlations between transdermal delivery of 5-aminolaevulinic acid and cutaneous protoporphyrin IX accumulation and effect of formulation. Br. J. Dermatol. 2002, 146, 853–862, doi:10.1046/j.1365-2133.2002.04715.x.
- O’Mahoney, P.; Khazova, M.; Eadie, E.; Ibbotson, S. Measuring Daylight: A Review of Dosimetry in Daylight Photodynamic Therapy. Pharmaceuticals 2019, 12, 143, doi:10.3390/ph12040143.
- Dirschka, T.; Ekanayake-Bohlig, S.; Dominicus, R.; Aschoff, R.; Herrera-Ceballos, E.; Botella-Estrada, R.; Hunfeld, A.; Kremser, M.; Schmitz, B.; Lübbert, H.; et al. A randomized, intraindividual, non-inferiority, Phase III study comparing daylight photodynamic therapy with BF-200 ALA gel and MAL cream for the treatment of actinic keratosis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 288–297, doi:10.1111/jdv.15185.
- Wiegell, S.R.; Haedersdal, M.; Philipsen, P.A.; Eriksen, P.; Enk, C.D.; Wulf, H.C. Continuous activation of PpIX by daylight is as effective as and less painful than conventional photodynamic therapy for actinic keratoses; a randomized, controlled, single-blinded study. Br. J. Dermatol. 2008, 158, 740–746, doi:10.1111/j.1365-2133.2008.08450.x.
- Yang, X.; Palasuberniam, P.; Kraus, D.; Chen, B. Aminolevulinic Acid-Based Tumor Detection and Therapy: Molecular Mechanisms and Strategies for Enhancement. Int. J. Mol. Sci. 2015, 16, 25865–25880, doi:10.3390/ijms161025865.
- Chaves, Y.N.; Torezan, L.A.; Niwa, A.B.; Sanches Junior, J.A.; Festa Neto, C. Pain in photodynamic therapy: Mechanism of action and management strategies. An. Bras. Dermatol. 2012, 87, 521–526, doi:10.1590/s0365-05962012000400001.
- Peng, Q.; Berg, K.; Moan, J.; Kongshaug, M.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy: Principles and experimental research. Photochem. Photobiol. 1997, 65, 235–251, doi:10.1111/j.1751-1097.1997.tb08549.x.
- Wiegell, S.R.; Wulf, H.C.; Szeimies, R.M.; Basset-Seguin, N.; Bissonnette, R.; Gerritsen, M.J.; Gilaberte, Y.; Calzavara-Pinton, P.; Morton, C.A.; Sidoroff, A.; et al. Daylight photodynamic therapy for actinic keratosis: An international consensus: International Society for Photodynamic Therapy in Dermatology. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 673–679, doi:10.1111/j.1468-3083.2011.04386.x.
- Morton, C.A.; Wulf, H.C.; Szeimies, R.M.; Gilaberte, Y.; Basset-Seguin, N.; Sotiriou, E.; Piaserico, S.; Hunger, R.E.; Baharlou, S.; Sidoroff, A.; et al. Practical approach to the use of daylight photodynamic therapy with topical methyl aminolevulinate for actinic keratosis: A European consensus. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1718–1723, doi:10.1111/jdv.12974.
- Wong, T.W.; Sheu, H.M.; Lee, J.Y.; Fletcher, R.J. Photodynamic therapy for Bowen’s disease (squamous cell carcinoma in situ) of the digit. Dermatol. Surg. 2001, 27, 452–456.
- Wang, B.; Shi, L.; Zhang, Y.F.; Zhou, Q.; Zheng, J.; Szeimies, R.M.; Wang, X.L. Gain with no pain? Pain management in dermatological photodynamic therapy. Br. J. Dermatol. 2017, 177, 656–665, doi:10.1111/bjd.15344.
- Babes, A.; Sauer, S.K.; Moparthi, L.; Kichko, T.I.; Neacsu, C.; Namer, B.; Filipovic, M.; Zygmunt, P.M.; Reeh, P.W.; Fischer, M.J. Photosensitization in Porphyrias and Photodynamic Therapy Involves TRPA1 and TRPV1. J. Neurosci. 2016, 36, 5264–5278, doi:10.1523/jneurosci.4268-15.2016.
- Borgia, F.; Giuffrida, R.; Caradonna, E.; Vaccaro, M.; Guarneri, F.; Cannavò, S.P. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12, doi:10.3390/biomedicines6010012.
- Mikolajewska, P.; Iani, V.; Juzeniene, A.; Moan, J. Topical aminolaevulinic acid- and aminolaevulinic acid methyl ester-based photodynamic therapy with red and violet light: Influence of wavelength on pain and erythema. Br. J. Dermatol. 2009, 161, 1173–1179, doi:10.1111/j.1365-2133.2009.09437.x.
- Ang, J.M.; Riaz, I.B.; Kamal, M.U.; Paragh, G.; Zeitouni, N.C. Photodynamic therapy and pain: A systematic review. Photodiagnosis Photodyn. Ther. 2017, 19, 308–344, doi:10.1016/j.pdpdt.2017.07.002.
- Grapengiesser, S.; Ericson, M.; Gudmundsson, F.; Larkö, O.; Rosén, A.; Wennberg, A.M. Pain caused by photodynamic therapy of skin cancer. Clin. Exp. Dermatol. 2002, 27, 493–497, doi:10.1046/j.1365-2230.2002.01065.x.
- Wulf, H.C. Daylight PDT acts by continuous activation of PpIX. Photodiagnosis Photodyn. Ther. 2019, 27, A1–A2, doi:10.1016/j.pdpdt.2019.04.012.
- McLellan, L.J.; O’Mahoney, P.; Logan, S.; Yule, S.; Goodman, C.; Lesar, A.; Fullerton, L.; Ibbotson, S.; Eadie, E. Daylight photodynamic therapy: Patient willingness to undertake home treatment. Br. J. Dermatol. 2019, 181, 834–835, doi:10.1111/bjd.17920.
- Rubel, D.M.; Spelman, L.; Murrell, D.F.; See, J.A.; Hewitt, D.; Foley, P.; Bosc, C.; Kerob, D.; Kerrouche, N.; Wulf, H.C.; et al. Daylight photodynamic therapy with methyl aminolevulinate cream as a convenient, similarly effective, nearly painless alternative to conventional photodynamic therapy in actinic keratosis treatment: A randomized controlled trial. Br. J. Dermatol. 2014, 171, 1164–1171, doi:10.1111/bjd.13138.