Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jing Zhao and Version 2 by Rita Xu.
Age-related macular degeneration (AMD) causes vision loss in the elderly population. Dry AMD leads to the formation of Drusen, while wet AMD is characterized by cell proliferation and choroidal angiogenesis. The retinal pigment epithelium (RPE) plays a key role in AMD pathogenesis. In particular, helioreceptor renewal depends on outer segment phagocytosis of RPE cells, while RPE autophagy can protect cells from oxidative stress damage.
- age-related macular degeneration
- autophagy
- oxidative stress
- retinal pigment epithelial cell
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of vision loss and irreversible blindness in the elderly worldwide; the number of people with AMD is expected to reach 288 million by 2040 [1]. Clinically, AMD is often classified as either dry or wet. Dry AMD, also known as non-neovascular AMD, is more common than wet AMD, accounting for 90% of total AMD cases. In dry AMD, pathological changes primarily occur in the retinal pigment epithelium (RPE) and Bruch’s membrane. Dry AMD progresses to a late stage of “geographic atrophy” characterized by the destruction and death of the RPE and photoreceptor cells. Chronic damage of the RPE leads to the formation of Drusen [2]. Wet AMD, also known as neovascular AMD, is characterized by choroidal neovascularization and an abnormal increase in vascular endothelial growth factor (VEGF) expression [3]. RPE cells are highly specialized epithelial cells with their apical surface in close contact with the outer segments of photoreceptors (POS) and the basal surface adherent to Bruch’s membrane. These cells play an important role in maintaining daily photoreceptor renewal and in the nutrition and function of the choriocapillaris [4]. Cells in the RPE phagocytose POS to ensure the continuous renewal of photoreceptor cells. At the same time, persistent photooxidative stress leads to the accumulation of reactive oxygen species (ROS). Under normal physiological conditions, the abundant antioxidants in RPE cells scavenge oxygen radicals to maintain homeostasis. In contrast, RPE cells in AMD exhibit increased levels of apoptosis, autophagy, and incomplete POS digestion, which in turn increase the burden of oxidative stress [5]. Persistent oxidative stress impairs the phagocytosis and autophagy of RPE cells, thereby increasing protein aggregation and activation of inflammatory vesicles.
Currently, most treatment options for AMD are administered to patients with advanced wet AMD, whereas no therapeutic agent can effectively prevent irreversible RPE and photoreceptor cell apoptosis and loss [6][7][6,7]. Indeed, an increasing body of evidence suggests that abnormal phagocytosis and autophagy of RPE cells contribute to AMD pathogenesis. Accordingly, therapeutics that target RPE cell phagocytosis and autophagy have been proposed as potential treatment options for AMD.
2. Pathogenesis of AMD
2.1. Risk Factors for AMD
Several blood vessels converge at the macula, which is therefore exposed to an abnormally high oxidative stress environment. Additionally, the RPE is a major source of ROS, owing to the abundance of mitochondria and extremely high metabolic activity [8][10]. The large amount of oxygen radicals that accumulates owing to persistent photooxidation has the potential to cause oxidative damage. Under normal physiological conditions, the antioxidant systems of RPE cells, such as superoxide dismutase, glutathione, and melanosomes, participate in scavenging oxygen radicals. AMD is associated with several environmental and genetic factors that increase the oxidative stress burden of the RPE [9][10][11,12]. Among these, age is a major risk factor for the development of AMD. With increasing age, the number of photoreceptors, ganglion cells, and RPE cells are reduced, and adverse reactions such as melanin loss, Bruch’s membrane thickening, and lipofuscin accumulation occur in RPE cells [11][13].The accumulation of proteins, lipids, and damaged DNA in aging tissues has a negative impact on the physiological function of cells. Consequently, the antioxidant function of RPE cells is compromised, making it incapable of removing the accumulated cell debris caused by the incomplete degradation of POS, resulting in a progressive increase in ROS. Thus, the increased sensitivity of aging cells to ROS aggravates oxidative stress. In addition, studies have found that age-related decreases in ubiquitin-dependent protein hydrolysis can lead to undegraded proteins forming basement deposits under the retina, which may contribute to the occurrence of AMD [12][13][14,15]. Smoking is considered to be the strongest non-genetic risk factor for AMD. Tobacco contains many strong oxidants, such as ROS, epoxides, and nitric oxide. Although the exact role that the oxidants in tobacco play in AMD is unknown, strong oxidants are known to deplete ascorbic acid and protein sulfhydryl groups in tissues, leading to the oxidation of DNA, lipids, and proteins [14][15][16][17][16,17,18,19]. In addition, obesity and a high-fat diet have recently been found to exacerbate subretinal inflammation and choroidal neovascularization and are the second most important risk factors for advanced AMD after smoking. A possible pathogenic mechanism is that a high-fat diet causes dysregulation of intestinal flora and impaired intestinal barrier function, which in turn leads to the release of microbial particles from the gut into the bloodstream. This induces the production of inflammatory factors and thus promotes chronic inflammation and pathological angiogenesis [18][19][20][20,21,22] (Figure 1).
Figure 1. Smoking, obesity, age, high-fat diet, arteriosclerosis, and exposure to sunlight are known major risk factors for age-related macular degeneration (AMD). Reactive oxygen species increase in the presence of these risk factors, inducing oxidative stress and leading to the accumulation of oxidized lipids, proteins, and DNA. This process gradually evolves into degeneration and necrosis of retinal pigment epithelial cells, eventually leading to AMD. The increase of ROS and the accumulation of oxidized lipids, proteins, and DNA are indicated by black arrows, while the progression of AMD is indicated by blue arrows. AMD, age-related macular degeneration; ROS, reactive oxygen species; RPE, retinal pigment epithelium.
2.2. RPE Cells in the Pathological Changes of AMD
The RPE is a part of the blood–retinal barrier, which consists of a single layer of RPE cells. Healthy RPE cells are hexagonal in shape, with tight connections forming between each RPE cell [21][23]. RPE cells have two types of microvilli that face the interphotoreceptor matrix and the photoreceptor outer segment [22][24]. Many organelles with specific functions can be found in RPE cells, including mitochondria, phagosomes, and lysosomes; these are located on the basal side of cells and participate in the renewal of phagocytes at the outer end of photoreceptors [23][25]. In humans, each RPE cell is responsible for the metabolism and renewal of 30–40 photoreceptor cells. The central fovea of the macula only has cone cells and RPE cells, and cone cells are the densest in the fovea. Consequently, the symptoms of impaired visual sensitivity and dark adaptation are particularly evident in patients with early AMD [24][25][26,27]. POS that are phagocytosed daily by RPE cells contain a large amount of unsaturated fatty acids, which are oxidized to produce a large amount of ROS during phagocytosis. As a result, the processes of continuous metabolism and renewal of the POS are the major sources of ROS in RPE cells [26][28]. The pathological changes that occur in early AMD are the degeneration of RPE cells and mesenchymal morphology, while photoreceptor function remains intact [27][28][29,30]. As the disease progresses, excessive metabolic wastes can no longer be degraded by autophagy and the intracellular lysosomal system of the RPE, which eventually leads to cell degeneration [13][15]. In AMD, the blood-retinal barrier weakens, lipofuscin is deposited, and failed lysosomes enlarge, as tight connections between cells separate, thin, or divide. During normal aging, Bruch’s membrane thickens. Nutrients and oxygen from the choroid and waste from the RPE must pass through Bruch’s membrane to maintain retinal homeostasis [29][31]. In the early stages of AMD, Bruch’s membrane thickens compared to normal aging. Electron microscopy revealed that abnormal deposits occurred above and below the RPE basement membrane in early and mid AMD, respectively, and may be involved in drusen formation. This process of change in Bruch’s membrane thickening reduces nutrient and substance exchange between RPE and choroid, leading to RPE photoreceptor dysfunction in AMD. As AMD progresses to advanced stages of the disease, Bruch’s membrane becomes thinner [30][32]. As one of the primary functions of the RPE is to maintain the function of photoreceptors, RPE cell degeneration leads to photoreceptor dysfunction and even death, resulting in AMD-related visual impairment. Therefore, it is speculated that RPE cells may develop AMD-related dysfunction if their phagocytic and autophagic functions are disrupted.3. Mechanism of RPE Cell Phagocytosis and Autophagy
3.1. Phagocytosis
Photoreceptors are susceptible to photooxidative damage, and their long-term health depends to a large extent on the processing of the aged portion of their outer segments. RPE cells are specialized for the phagocytosis of POS and are the only means of removing their aged portion, a process also known as heterophagy [31][33]. Each RPE cell is in close contact with approximately 30 photoreceptor (cone and rod) cells, which they will phagocytose and then remove their outer segments. Notably, the shedding and phagocytosis of POS are regulated by circadian rhythms, with most occurring soon after the onset of light [32][34]. The tip of POS contains high concentrations of free radicals, photodamaged proteins, and lipids. POS is maintained at a constant length through continuous maintenance of the balance between the shedding of POS tips and the formation of new POS. Exposed phosphatidylserine gradually accumulates at the POS tip under light exposure [33][35]. Photoreceptor cells are in contact with the RPE in a fixed manner, and the apical microvilli of RPE cells wrap around the POS before phosphatidylserine accumulation [34][35][36,37]. Phagocytosis of POS can be divided into four distinct phases: recognition and binding of the POS, POS ingestion, phagosome formation, and phagosome digestion. In the first step of phagocytosis, a receptor tyrosine kinase (MerTK) mediates the specific binding of POS to the RPE membrane, which in turn activates a second messenger that transduces the signal to the cytosol [36][38]. Notably, the αvβ5 integrin receptor is essential in the POS binding process. MerTK itself does not bind directly or indirectly to actin but requires the αvβ5 integrin receptor to provide traction for mechanical attachment to actin. The αvβ5 integrin receptor itself is triggered by MerTK [37][39], and so these two processes interact with each other, mediated by the activation of focal adhesion kinase [38][39][40][40,41,42]. Therefore, cells lacking the MerTK receptor can bind to but not ingest POS [31][33] (Figure 2). The maturation of phagosomes and their fusion with lysosomes occur during the second half of heterophagy in RPE cells. Cathepsins are proteases that degrade proteins. Currently, the primary role of cathepsin D (CTSD) in RPE cells has been identified as the degradation of POS into glycopeptides in lysosomes. A study found that mice lacking CTSD develop retinal degeneration [41][42][43,44]. Additionally, cystatins are inhibitors of lysosomal cysteine proteases highly expressed in RPE cells, and mutations in cystatin genes have been shown to be closely associated with RPE degeneration and AMD pathogenesis [43][44][45,46]. A recent study identified βA3/A1-crystallin as a novel lysosomal component in the RPE that regulates phagocytosis and autophagy [45][47].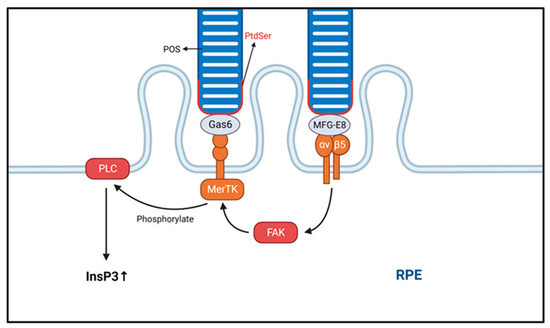
Figure 2. The phagocytic process of the photoreceptor outer segment (POS) is shown. The process of POS ingestion is transduced by integrins. The transduction of intracellular signals begins with the specific binding of macrophage c-mer tyrosine kinase (MerTK). Eventually, the intracellular inositol-1,4,5-trisphosphate (InsP3) content is increased, and InsP3 activation promotes POS ingestion. Integrin αvβ5 binds to phosphatidylserine (PtdSer) via MFG-E8, while milk fat globule-epidermal growth factor (EGF) factor 8 (MerTK) binds via growth arrest-specific protein 6 (Gas6). POS, photoreceptor outer segment; RPE, retinal pigment epithelium.
3.2. Autophagy
RPE cells are not only one of the most active phagocytic cells in the body but are also extremely metabolically active post-mitotic cells with high rates of autophagy. Mammalian cells often exhibit three types of autophagy: microautophagy, chaperone-mediated autophagy, and macroautophagy. Macroautophagy is the most common autophagy in RPE cells and differs from the other two types in that it depends on the formation of autophagosomes. The contents of autophagosomes are degraded by lysosomal enzymes after fusion with lysosomes, whereas, in microautophagy and chaperone-mediated autophagy, the damaged proteins, lipids, and organelles are directly transported to lysosomes. Impaired autophagy in RPE cells can lead to the accumulation of damaged proteins, which eventually leads to cell degeneration and death.3.2.1. Autophagy Mechanism
Autophagy is a highly complex intracellular garbage removal pathway, the first step of which includes autophagosome formation. Autophagosomes comprise double-membrane vesicles that participate in the clearance of damaged or nonfunctional organelles, proteins, and other substances [48][50]. Autophagosome formation requires the participation of a series of autophagy-related genes and proteins, including proteins from the ATG (autophagy-related genes) family and LC3 (microtubule-associated protein 1 light chain 3). Autophagosomes are transported throughout the cell via microtubule dynamics to allow them to fuse with lysosomes. Subsequently, the autophagosome contents are degraded by acid hydrolase, proteases, and other enzymes, allowing them to be recycled. Initiation and activation of the autophagy pathway are highly regulated processes, regulated by myriad signaling pathways and molecules, including mTOR (mammalian target protein of rapamycin), AMPK (adenosine 5‘-monophosphate (AMP)-activated protein kinase), and ULK1/2 (unc-51 like autophagy activating kinase) [49][50][51,52] (Figure 3).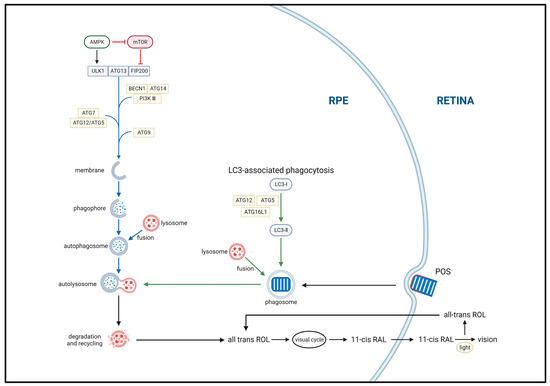
Figure 3. Two main autophagy pathways associated with the classical visual cycle of retinoids. The ULK1, ATG13, and FIP200 complex drives autophagy initiation. Autophagosome formation is primarily mediated by the BECN1, ATG14, and PI3K III complex with double-membrane structures provided by ATG9 vesicles. Autophagosome elongation and closure are coupled through the ATG5 and ATG15 complex. mTOR and AMPK are autophagy inhibitors and inducers, respectively. Autophagy begins with the sequestration of aging, damaged, or unnecessary cellular components, such as organelles and proteins, by double-membrane structures, forming phagophores (autophagosomes). These structures then fuse with lysosomes to form autolysosomes, where the enclosed materials are degraded by acid hydrolases and proteases, and the resulting breakdown products are released into the cytoplasm for recycling. Additionally, LC3-associated phagocytosis (LAP)—a noncanonical form of autophagy—is responsible for the degradation of photoreceptor outer segments (POS) and completion of the retinoid visual cycle. Upon initiation of phagocytosis, LAP immediately recruits ATG12, ATG5, and ATG12L1 to form a complex, as well as LC3 for lipidation. Following POS degradation, all-trans-retinol is converted to 11-cis-retinol in the RPE and transported out of the cell, completing the classical visual cycle of retinoids.