1. Introduction
According to World Health Organization (WHO) global health estimates, cardiovascular diseases (CVDs) remain the leading cause of morbidity and mortality in the world, claiming 17.9 million lives each year
[1]. CVDs are disorders related to the heart and circulatory system, and ischemic heart disease is the most frequent, causing an estimated annual mortality of nine million people. CVDs mainly affect men over 40 years and older adults, with a significant increase over 70 years of age; it is known that three-quarters of deaths belong to low- and middle-income countries
[1]. In Latin America, CVD represents 2 million deaths and ischemic heart disease 73.6 deaths per 100,000 people per year
[2]. During ischemia, due to the intimate relationship between oxygen supply and coronary blood flow, normal perfusion to the cardiomyocyte is interrupted, leading to the termination of aerobic metabolism, creatine phosphate depletion, and anaerobic glycolysis, causing mitochondrial dysfunction
[3]. Mitochondria not only produce adenosine triphosphate (ATP), but also participate in pivotal metabolic processes and cell signaling. This is the reason why mitochondrial dysfunction results in low levels of ATP, energy stress, production of reactive oxygen species (ROS), oxidative stress and alteration in the ionic balance, mainly in calcium homeostasis
[4].
Metformin, a common and affordable drug for hyperglycemia, has been shown to exert cardioprotective
[5][11] and anti-inflammatory effects beyond glycemic control
[6][7][8][12,13,14]. Metformin is a biguanide drug used as the first-line medication in the treatment of type 2 diabetes
[6][12], and has been documented to inhibit complex I of the mitochondrial respiratory chain, leading to activation of the cellular energy sensor AMP-activated protein kinase (AMPK). AMPK is activated by increasing adenosine monophosphate (AMP), ATP and adenosine diphosphate (ADP)/ATP ratios. Activated AMPK restores energy homeostasis by turning on catabolic pathways generating ATP while switching off cellular processes consuming ATP
[7][13]. AMPK plays an important role in numerous diseases, such as type 2 diabetes mellitus (DM), dyslipidemia, and non-alcoholic fatty liver, as well as control of infections such as tuberculosis, ischemia, inflammation, and the aging processes
[9][15]. Evidence has identified the fact that metformin may have many other benefits, including protection against cardiac ischemia-reperfusion (I/R) injury, atherosclerosis, inflammation, oxidative stress, and reducing the risk of stroke and death caused by CVD
[6][10][11][12][13][14][12,16,17,18,19,20].
The potential cardioprotective effect of metformin may extend beyond its AMPK-mediated effect
[15][21]. Evidence has shown that the administration of metformin during reperfusion leads to the activation of several kinases of the Reperfusion Injury Salvage Kinase (RISK) pathway, in which phosphatidylinositol-3-kinase (PI3K) and Protein kinase B (Akt) are involved
[16][17][18][22,23,24]. Similarly, it has been reported that activation of multiple RISK pathway protective kinases inhibits the opening of the mitochondrial permeability transition pore (mPTP) during reperfusion, thereby limiting the size of myocardial infarction (MI)
[19][25]. Also, metformin inhibits cell death by suppressing inflammation during apoptosis, and protects against myocardial I/R injury by activating several pathways that end up controlling and inhibiting autophagy and regulating apoptosis. This cardioprotective effect is linked to Akt signaling pathway activation
[19][25].
2. Cardioprotective Effects of Metformin on Myocardial I/R
In addition to being a popular antidiabetic, the literature suggests that there is an important cardioprotective effect in the use of metformin. Numerous preclinical studies have evaluated the effects of metformin on I/R-induced cell death and inflammation
[20][21][22][23][59,60,61,62] (
Figure 1).
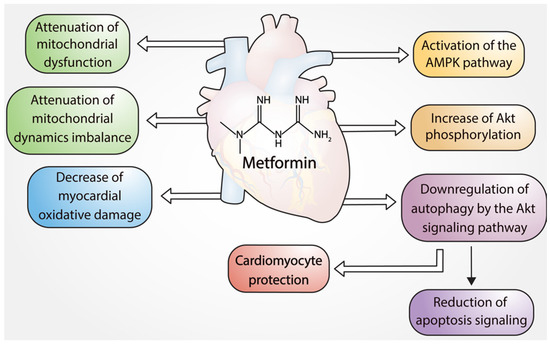
Figure 1.
Cardioprotective effects of metformin.
2.1. Cardioprotective Effects of Metformin via Inhibition of Apoptosis
Palee et al. 2020 demonstrated in an experimental study that metformin treatment is cardioprotective in the setting of myocardial I/R injury, by attenuating mitochondrial dysfunction, mitochondrial dynamic imbalance, and apoptosis
[23][62].
Some cardioprotective effects of metformin are mediated for AMPK. This kinase plays an important role in regulating myocardial energy metabolism, and reducing of I/R injury
[23][24][25][62,63,64]. A recent experimental in vitro study performed by Zhang et al. showed that AMPK activation with metformin protected against apoptosis induced by cardiac I/R injury. In isolated rat hearts subjected to I/R, metformin reduced the size of the infarct and inhibited cardiac fibrosis through the reduction of proinflammatory cytokines (TNF-α, IL-6, IL-1β) and the suppression of the activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome
[5][11]. Shi et al. showed in an experimental study that metformin reduced the oxidative stress injury induced by myocardial I/R. Metformin upregulated the phosphorylation of AMPK and decreased the NOX4 gene expression, leading to the decrease in myocardial oxidative damage and apoptosis, thereby alleviating reperfusion injury
[25][64]. Metformin has also been reported to protect cardiomyocytes against H
2O
2-induced apoptosis through the AMPK/CCAAT-enhancer-binding protein (C/EBP) beta/miR-1a-3p/grpq4 (β/miR-1a-3p/GRPQ4) protein pathway. Through this pathway, Zhang et. al., determined that the expression of miR-1a-3p was significantly increased in neonatal rat ventricular cells which were exposed to H
2O
2 in vitro and in the hearts of mice that suffered I/R-induced injury. The miR-1a-3p follows protein GRP94, which results in the accumulation of unfolded or misfolded proteins, generating ER stress. C/EBP β directly induces the upregulation of miR-1a-3p, by binding to its promoter. Metformin appears to activate AMPK and at the same time significantly reduce the levels of C/EBP β and miR-1a-3p, compared to those of the control group in the study
[26][65]. These in vitro and in vivo results confirm the positive effect of metformin for the treatment of cardiac I/R injury by attenuating cell apoptosis.
2.2. Cardioprotective Effects of Metformin on I/R Injury via Modulation of ROS
Metformin can additionally prevent I/R injury through other mechanisms such as modulation of ROS levels and oxidative damage during reperfusion. Metformin treatment reduced 4-HNE (4-hydroxynonenal) staining, an indicator of lipid peroxidation, after I/R injury
[25][64]. This result is consistent with decreased NOX4 levels in metformin-treated hearts, which were dependent on AMPK activation. Interestingly, Asensio-Lopez et al., claimed that metformin protective effects over myocardial infarction, involved the participation of a mitochondrial NAPDH oxidase 4 (
mitoNox), a mitochondria-localized NOX4
[27][66], which has been associated with increased ROS production during I/R and adverse myocardial remodeling (PMID: 25589557). Additionally, metformin has been shown to modulate ROS levels in I/R injury, through upregulation of antioxidant enzymes, such as MnSOD (manganese-containing superoxide dismutase)
[10][16]. Similarly to other metformin effects, MnSOD upregulation was an AMPK-dependent process
[10][16]. Thus, metformin has cardioprotective effects, involving oxidative stress management via AMPK activation, through reduction of pro-oxidant enzymes like (mito)NOX4, and upregulation of antioxidant enzymes such as MnSOD
[25][64].
2.3. Cardioprotective Effects of Metformin on I/R Injury via Autophagy
Autophagy is an important physiological process in cells, which can degrade dysfunctional organelles and proteins. Autophagy plays a double role in myocardial I/R injury, and depends on the stimulus conditions. Moderate levels of autophagy can exert a protective effect against oxidative stress and the first minutes of hypoxia
[25][64]; however, excessive activation of autophagy can cause cell death. Studies by Huang et al. and Wu et al. are an example of this double and opposite role of autophagy during I/R. Huang et. al. showed a destructive role of autophagy in myocardial I/R injury, and discovered that metformin inhibits apoptosis and inflammation during I/R via the restoration of autophagosome processing. Metformin induced the phosphorylation of Akt, which activates the mammalian target of rapamycin (mTOR), an inhibitor of autophagy. The density of autophagosomes in metformin-treated mice was much lower than in the control group, after I/R injury
[26][65]. On the other hand, Wu et al. found that metformin improved cardiac function in infarcted rats previously treated with this drug, as evaluated by echocardiography, and promoted myocardial autophagy in in vivo models, thus revealing reductions in apoptosis and areas of infarction through the promotion of autophagy. Metformin promoted autophagy by increasing the protein expression of light chain 3 (LC3)-II, autophagy related (ATG) 5, ATG7 and Beclin1, and through the AMPK pathway during MI
[28][67].
Fei et al. 2020 showed in their experimental study that metformin treatment in mice subjected to acute MI activated the autophagic flux in macrophages. Metformin decreased the expression of NLRP3 and p62 and increased the ratio of LC3II/LC3I. In addition, metformin treatment abrogated the effects of hydrogen peroxide and a combination of mitochondrial DNA (mtDNA) and ATP over the expression of NLRP3, and cleaved caspase-1 as well as intracellular ROS production in RAW264.7 macrophages. The suppression of NLRP3 by metformin was mainly attributed to the activation of the autophagy
[29][68].
Metformin plays a dual role regarding autophagy in cardiomyocytes; it usually activates autophagy via AMPK or inhibits it in some models, via Akt/mTOR
[19][25]. However, it is pertinent to consider that this difference may be due to the experimental conditions and models used. In fact, the role that autophagy plays in the context of cardioprotection is controversial; however, more studies are required to clarify whether autophagy is crucial in the cardioprotective mechanism of metformin.
Zhang et al. showed this dual action in an experimental study in murine models in which I/R myocardial damage was induced by transient occlusion of the left anterior descending artery. In this experimental model, the specimens were treated with metformin, showing a reduction in the ischemic area mediated by autophagosomes. The authors detected that the proteins associated with the membrane of the autophagosome LC3-II, BECLIN-1 and ATG5 were significantly increased in the group that received metformin; also shown was the increase in the expression of p-AMPK and the inhibition of p-mTOR, which represents an activation of autophagy through the regulation of this pathway with a partial reduction in myocardial damage and the subsequent cardioprotection
[30][69].
2.4. Cardioprotective Effects of Metformin via Mitochondrial Function
Mitochondrial oxidative metabolism is the key source of cardiac energy, and mitochondrial dysfunction is considered the main myocardial mechanism that leads to a contractile failure after I/R injury. This organelle preserves its homeostasis and membrane potential through the mPTP, which is compromised by the irreversible myocardial damage that follows I/R injury, so there is a close relationship between both elements. Therapeutic strategies targeting I/R injury and also targeting mPTP inhibition have been developed to modulate energy homeostasis, mitochondrial function, and ROS production in cardiomyocytes. Specifically, it has been described that the translocator protein (TSPO) is the key component of mPTP that modulates these changes. Upregulation of TSPO expression was found to be associated with ROS accumulation and disruption of mitochondrial homeostasis. Myocardial TSPO expression was measured serially in correlation with the degree of mitochondrial homeostasis and cardiac function in a rat model exposed to myocardial I/R injury, using positron emission tomography imaging. The expression of TSPO was correlated with inflammatory changes in the infarcted area, and was associated with an upregulation of p-AMPK/AMPK; however, these effects were reversed with the use of metformin
[31][70].
Aging has implications in the weakening of mitochondrial function, predisposing a greater cardiac injury during I/R. Stress on the cardiac endoplasmic reticulum (ER) increases with age, contributing to mitochondrial dysfunction. A study was conducted with mixed young (3 months) and old (2 years) mice; one group received metformin and sucrose water, while another group received sucrose alone, for 2 weeks. Cardioprotection was assessed using an isolated rat heart subjected to 25 min global ischemia and 60 min reperfusion, subsequently measuring the infarct size area. Factors highly involved in ER stress such as C/EBP homologous protein and cleaved activated transcription factor 6, were reduced in 24-month-old mice treated with metformin, compared to mice the same age that only received sucrose, which translates into a reduction in ER stress. In addition, metformin-treated rodents were found to have smaller infarct size after I/R
[32][71].
In another experimental study, metformin treatment improved the mitochondrial respiratory function and mitochondrial membrane potential in male C57/BL6 mice subjected to myocardial infarction. The effects of metformin were evaluated during 8 weeks after MI, and this drug upregulated the expression of sirtuin (Sirt3) and the activity of peroxisome proliferator-activated receptor Γ coactivator 1 alpha (PGC-1α) in myocardial tissue of heart failure. Metformin decreased the acetylation of PGC-1α through the up-regulating of Sirt3 and thus mitigated the mitochondrial damage
[33][72]. Metformin also attenuated I/R injury by decreasing mitochondrial dynamic imbalance and reducing cardiac mitochondrial dysfunction due to downregulation of ROS production, mitochondrial membrane depolarization, and swelling of the mitochondria
[26][65].
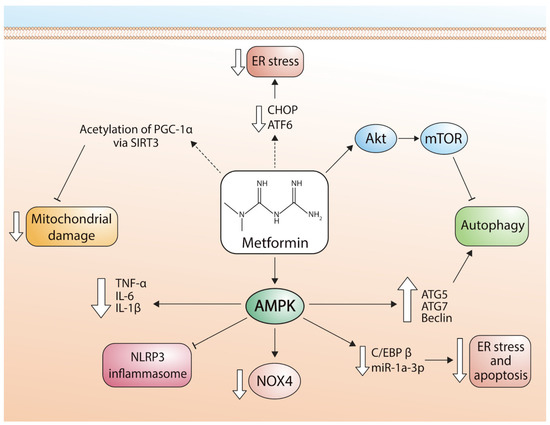
Figure 2 describes the signaling pathways used by metformin to regulate mitochondrial function.
Figure 2. Pathways involved in the cardioprotective effects of metformin. ER stress: endoplasmic reticulum stress; CHOP: transcription factor CCAAT-enhancer-binding protein homologous protein; ATF6: activating transcription factor 6; Akt: alpha serine/threonine-protein kinase; mTOR: mammalian target of rapamycin; PGC-1α: peroxisome proliferator-activated receptor-gamma coactivator; SIRT3: Sirtuin-3; TNF-α: tumor necrosis factor alpha; IL-6: Interleukin 6; IL-1β: Interleukin 1 beta; NLRP3: NLR family pyrin domain containing 3; NOX4: NADPH oxidase 4; C/EBPβ: CCAAT/enhancer-binding protein beta; miR-1a-3p: microRNA-1 family; ATG5: autophagy related 5; ATG7: autophagy related 7.
These preclinical findings support the ability of metformin to promote cardioprotection by reducing mitochondrial dysfunction, dynamic mitochondrial instability, and apoptosis, which decrease cardiac tissue injury. Therefore, it is plausible to postulate a potential clinical benefit of acute treatment with metformin in acute myocardial infarction.
Figure 2 shows the different pathways by which metformin exerts cardioprotective effects in cardiomyocytes via regulation of apoptosis, autophagy, and mitochondrial function (Figure 2).
2.5. Effects of Metformin on Cardiac Function after I/R Injury
The protective mechanism of metformin in cardioprotection is not fully understood. Nonetheless, several studies have investigated the effect of metformin during and after episodes of induced I/R. Su et al. (2022) developed an assay using murine models to determine the mechanism by which metformin attenuates cell damage caused by ischemia and subsequent reperfusion. They used rats subjected to occlusion of the left anterior descending coronary artery. The metformin group was treated with metformin twice a day for 30 days from the first day after the operation, while the control group was treated with saline solution for 30 days. The MI and fibrosis in the central ischemic area of the control group were more severe than those in the metformin group. The authors found that one month after the start of the intervention with metformin, the severity of the I/R event decreased, and ventricular remodeling was delayed. This study reported, for the first time in an in vivo molecular imaging assay, that metformin treatment improved glucose metabolic activity in ischemic myocardium and had a myocardial protective effect at the molecular level by delaying the development of ventricular remodeling triggered by I/R injuries
[34][73].
Although more studies are needed to fully elucidate the action of metformin as a cardioprotective agent, current studies highlight how this drug protects cardiac function during myocardial injury. Jo et al. (2020) used murine heart models to demonstrate the effectiveness of metformin on left ventricular (LV) diastolic function using echocardiography in a rat myocardial I/R injury model. The model of myocardial I/R injury in rats involved tying off the left anterior descending coronary artery for 30 min, which resulted in MI-confirmed echocardiography
[35][74].
Jo et al. (2020) confirmed the positive effect of metformin on the early stage of MI in rats through various assessments such as total weight change, relative heart weight, LV systolic and diastolic function using echocardiography on days 1, 3, and 7, and the degree of fibrosis. The LV diastolic dysfunction during MI is an important indicator of poor surgical outcomes and recurrence in human MI patients. The MI group exhibited decreased ejection fraction (EF) and fractional shortening values, reflecting LV systolic dysfunction, and also showed reduced E’ values (echocardiogram E wave) and increased E/E’ values; the E/e’ ratio refers to the relationship between the velocity of blood flow into the heart’s left ventricle during early diastole and the velocity of the mitral valve annulus during the same phase, reflecting LV diastolic dysfunction, which resembles human MI. The elevation of LV filling pressure is the key indicator of poor outcomes in human MI patients. This study also examined gene expression profiles, to provide a molecular basis for understanding the mechanism of action of metformin in this model
[35][74]. The findings suggest that metformin can attenuate LV diastolic dysfunction induced by MI, and the molecular basis for this effect may involve alterations in immune/inflammation and cardiovascular system pathways, as well as fatty acid metabolism, mitochondrial biogenesis, and transforming growth factor-beta/bone morphogenetic protein and Janus kinase/signal transducers and activators of transcription signaling pathways. Overall, these results provide important insights into the potential clinical use of metformin in the treatment of MI, and highlight the need for further investigation of its effects on cardiovascular function
[35][74].
Palee et al. (2020) investigated the efficacy of metformin in providing cardioprotection in a rat model of cardiac I/R injury. The results showed that metformin administration prior to cardiac I/R injury attenuated mitochondrial dysfunction, dynamic imbalance, and apoptosis, leading to a decrease in infarct size and improvement in cardiac function. The study also found that the optimal dose of metformin was 200 mg/kg, although doses of 100 and 400 mg/kg also provided some cardioprotective effects. These findings suggest that acute treatment with metformin may have clinical benefits for patients with MI. The reduction in excessive mitochondrial fission is important, as it is associated with cardiomyocyte apoptosis during cardiac I/R injury. The study also found that the reduction in phosphorylation of the cardiac gap junction protein Cx43 at serine 368 by metformin may have contributed to the higher mortality rate observed with a higher dose of metformin. Furthermore, the optimal dose and timing of metformin administration in the context of clinical relevance should be determined in future studies
[36][75].
Eppinga et al. (2017) described in a randomized clinical trial the effects of metformin on the metabolic profiles of non-diabetic myocardial infarction patients and identified the prognostic metabolites predicting left ventricular ejection fraction (LVEF) and infarct size 4 months post-MI. Metformin treatment resulted in higher alanine levels and lower phospholipid content of large high-density lipoprotein (HDL) particles, as compared to controls. Higher triglyceride levels in HDL and several HDL subfractions measured 24 h post-MI were associated with favorable outcomes in terms of higher LVEF and smaller infarct size four months post-MI. The researchers also found that decreased HDL triglyceride levels measured 24 h post-MI predicted higher infarct size. The authors observed beneficial effects of higher levels in HDL measured 24 h post-MI on infarct size and LVEF
[37][76].
The use of metformin in the management of cardiovascular disease has shown promising results, particularly in the setting of acute myocardial infarction. The cardioprotective effects of metformin may be attributed to its ability to modulate various metabolic pathways involved in energy metabolism, inflammation, and oxidative stress. Studies have demonstrated that the early administration of metformin following acute MI improves LV EF and reduces infarct size. Additionally, metformin has been shown to alter lipid profiles and increase the levels of cardioprotective HDL subfractions
[37][76]. These findings suggest that metformin could be a valuable adjunct therapy in the management of acute MI and the prevention of recurrent cardiovascular events. However, further studies are required to establish the optimal dosing and duration of metformin therapy in different patient populations and to elucidate the underlying mechanisms of its cardioprotective effects.