Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by David Martín-Hernández and Version 3 by Rita Xu.
Sphingosine-1-phosphate (S1P) is a bioactive lipid that regulates both the immune response and the permeability of biological barriers. Notably, S1P-based drugs, such as fingolimod and ozanimod, have received approval for treating multiple sclerosis, an autoimmune disease of the central nervous system (CNS), and ulcerative colitis, an inflammatory condition of the colon, respectively.
- sphingosine-1-phosphate
- psychiatric diseases
- immune system
- inflammation
- barrier function
1. Immune Dysregulation and Barrier Function in Psychiatric Diseases
1.1. Immune Dysregulation in Psychiatric Diseases
A substantial proportion of psychiatric patients (20–60%) fail to achieve a complete remission with currently available treatments, a statistic that remains consistent for severe mental illnesses (SMI), including major depressive disorder (MDD) and schizophrenia (SZ) [1]. This situation has prompted the scientific community to explore new pathways in the pathophysiology of these SMI, which may unveil new targets, diagnostic tools, or risk biomarkers to personalize and stratify treatments. In this context, a promising area of research in recent years has revolved around the potential dysregulation of immune or inflammatory responses in both the brain and the rest of the body in these diseases. Evidence from both preclinical models and studies involving human samples suggests a pathophysiological role of these processes. Importantly, therapeutic interventions could potentially target these pathways to enhance patient treatment outcomes.
Neuroinflammation is a common feature of many neurodegenerative diseases, and over the last two decades, this concept has also extended to psychiatric disorders. To illustrate this, researchwers will go into details with MDD and SZ, but immune alterations have been also found in other mental pathologies, including such as bipolar disorder (BD) [2], autism spectrum disorder (ASD) [3], attention deficit hyperactivity disorder (ADHD) [4], and eating disorders (EDs) [5].
The first genome-wide association studies (GWAS) [6] identified a substantial number of genes (108) implicated in cohorts of approximately 40,000 patients with SZ. Several other genetic association studies have validated these data. Among these genes, a prominent cluster (the largest, surpassing other neurotransmitter-related ones) located on chromosome 6 may regulate the inflammatory and immune response. This discovery has confirmed previous clinical evidence and underscored the connection between the immune system and SZ, a relationship further corroborated in MDD [7][8][7,8].
Among the nonspecific inflammatory features identified in blood analyses of psychiatric patients, leukocytosis with neutrophilia is one of the most consistent in first-episode psychosis, SZ, and BD [9][10][9,10]. Another nonspecific inflammatory parameter elevated in SZ patients is the erythrocyte sedimentation rate [11]. Interestingly, longitudinal studies encompassing tens of thousands of individuals tracked over a period of 45 years (who had their erythrocyte sedimentation rate measured during health examinations before military service), revealed that those who later developed psychotic symptoms or SZ exhibited higher levels of this nonspecific inflammatory parameter years earlier. This suggests a potential sensitizing effect of prior low-grade inflammatory stimulus in vulnerable individuals.
Hence, innate and adaptive immune alterations have been observed in MDD [12][13][14][12,13,14] and SZ [15][16][17][15,16,17]. Moreover, some studies support the existence of psychiatric and autoimmune comorbidities [18]. Imaging studies (in vivo, positron emission tomography, or postmortem tissue evaluation for anatomopathological assessment) have revealed microglial activation in the brains of patients with MDD [19][20][19,20] or SZ [21][22][21,22]. Interestingly, there is a correlation between systemic inflammation and brain functional consequences: patients with higher plasma C-reactive protein (CRP) levels showed fewer connectivity indices in the central nervous system (CNS) [23].
It is well-established that certain immunomodulatory treatments, such as interferon therapy, may induce MDD symptoms [24]. Recent evidence also indicates the development of SZ-related behaviors under similar circumstances [25]. Similarly, the administration of cytokines or an immunological challenge has caused behavioral changes. For instance, in humans, injection of lipopolysaccharide (LPS), the main component of the outer membrane of Gram-negative bacteria, induces sickness behavior (depressed mood, anhedonia, anxiety, etc.), systemic cytokine increases, and microglial activation [26]. Behavioral alterations resembling MDD or SZ, triggered by various immune stimuli—such as LPS and polyinosinic: polycytidylic acid (Poly I:C)—have served to develop preclinical models for the study of these pathologies [27].
Given the existing evidence, there exists a bidirectional relationship between the immune system and current pharmacological treatments: antidepressants and antipsychotics have demonstrated anti-inflammatory effects beyond their conventional neurotransmitter actions [28][29][28,29], while anti-inflammatory drugs have shown antidepressant and antipsychotic effects or the ability to enhance the efficacy of these drugs when used as adjuvants [30][31][32][30,31,32]. Nevertheless, the question of whether low-grade inflammation or immune dysregulation is a cause or consequence of mental health disorders remains an area of ongoing debate.
The study by Kappelmann et al. [11], mentioned earlier, suggested a potential causal link in individuals entering military service. Moreover, a long-term study in the Avon County of the United Kingdom has reported an “imprinting” effect [33]. Children exhibiting higher levels of IL-6 at the age of 9 were at increased risk of developing psychosis or psychotic symptoms by the age of 18. In another study conducted in Denmark, involving nearly 80,000 individuals followed over several years, patients with SZ or psychotic symptoms displayed elevated inflammatory parameters, such as C-reactive protein (CRP). Specifically, individuals in the fourth quartile of CRP levels had the highest likelihood of developing the disease. These findings hold considerable statistical robustness, as the difference persists in sub-analyses adjusted for sex and age, and also in multivariate analyses [34]. However, the relationships with depressive disorders are not as clearly defined.
The possibility of a widespread inflammatory environment throughout the system reaching the brain via a more permeable blood–brain barrier (BBB) remains an intriguing open question in numerous ongoing research projects worldwide. The origin of these inflammatory elements in circulation is still a topic of debate, whether they stem from a mobilization from cellular nests or from the activation of immune cells by stimulating molecules. In this regard, recent preliminary data indicate that patients with SZ or MDD showed an increase in the permeability of the intestinal barrier [35][36][35,36] and vice versa, patients with inflammatory bowel diseases were susceptible to psychiatric symptoms through leaky gut, a phenomenon related to increased intestinal permeability [37]. Moreover, recent GWAS revealed a shared genetic background between psychiatric and gastrointestinal disorders [38]. This concurrence of phenomena may signify the potential translocation of bacteria-derived LPS from the colon, as demonstrated in preclinical settings using stress protocols [39].
Studies by Cai et al. [40] have recently shown that LPS can reach the BBB in mental disorders. Specifically, there is an increase in perivascular BBB macrophages in postmortem specimens, along with elevated inflammation markers, such as vascular cell adhesion molecule (VCAM), identified in perivascular areas and astrocytes. Consequently, both the brain and intestinal barriers emerge as crucial structures in psychiatric diseases, and their dysfunctions may play a pivotal role in driving their pathophysiology.
1.2. The Brain Barriers in Psychiatric Diseases
1.2.1. Structure and Function of the Brain Barrier
The conventional notion of a singular BBB has evolved into the recognition of several distinct brain barriers, each occupying specific neuroanatomic locations and comprising unique structural components that regulate interactions between brain parenchyma and blood-born molecules or cells [41][42][41,42]:
-
BBB: constituted by microvascular endothelial cells with apical tight junction complexes lining the cerebral capillaries that traverse the brain and spinal cord. Heterogenic endothelial cells shape the neurovascular complex interacting with neurons, astrocytic endfeet, myocytes, perivascular macrophages, pericytes, and extracellular matrix components [43].
-
Arachnoid (or meningeal) barrier: formed by the avascular arachnoid epithelium, featuring tight junctions as a barrier between the cerebrospinal fluid (CSF)-filled subarachnoid space and other meningeal structures.
-
Blood-CSF barrier: this barrier, also constituted by epithelial cells with apical tight junctions, separate the blood vessels of the choroid plexus (fenestrated and leaky) from the CSF.
-
The fetal CSF-brain barrier: during early development, this barrier exists between the CSF and brain parenchyma during early development. It acts as a functional barrier when junctions interconnect ependymal cells.
-
The adult ventricular ependyma: as development progresses, the ependymal cells lose their ability to restrict the passage of larger molecules, such as proteins, between the CSF and the brain.
There are additional brain-blood interfaces known as the circumventricular organs (CVOs), which are characterized by the absence of the BBB and are highly vascularized with fenestrations [43]. These brain structures, located in the third and fourth ventricles, can classify as sensory (subfornical organ, organum vasculosum of the lamina terminalis, and area postrema) or secretory (subcommissural organ, pituitary neural lobe, median eminence, and pineal gland) CVOs [43].
Different types of junctions participate in connecting endothelial cells of the BBB. Intercellular tight junctions (TJs) provide firm mechanical stability with low transcytosis rates and composed of proteins spanning the intercellular gap (occludins and claudins), junctional adhesion molecules (JAMs), and regulatory proteins (Zonula Occludens 1, 2, 3 (ZO-1, ZO-2, ZO-3)), responsible for linking transmembrane proteins with the actin cytoskeleton [44]. Adherens junctions consist of cadherins and catenins that link the actin cytoskeleton with cadherins, while gap junctions encompass tissue-specific isomers of the connexin (Cx) family. Collectively, these junctional proteins ensure strict regulation of paracellular permeability in the BBB [45].
Transporters are another vital component of the BBB structure, regulating the transport of nutrients, neuroactive peptides, large proteins, and other molecules into and out of the brain through multiple mechanisms, including carrier-mediated transport (facilitated diffusion), receptor-mediated transport systems, and active efflux transporters (ATP-binding cassette (ABC) transporter superfamily [46]. Transcytosis, another transport mechanism across the brain endothelium, relies on vesicle trafficking and is strictly controlled under physiological conditions. Three types of transcytosis regulate the transcellular transport of lipophobic molecules across the cerebral endothelium: fluid phase, absorptive, and receptor-mediated (clathrin and caveolae dependent) [47]. Lastly, another important transport mechanism associated with the BBB is the glymphatic system, which primarily facilitates debris clearance by controlling the exchange of CSF and interstitial fluid through astrocytic cells [48].
1.2.2. Brain Barrier Dysfunction in Psychiatric Diseases
One of the hallmarks of neuroinflammation is the dysregulation in the structure and function of brain barriers, leading to a loss of barrier integrity and increased general permeability to molecules and cells. Dysfunction of the BBB is a crucial event in the pathophysiology of neurological conditions such as traumatic brain injury or stroke [49][50][49,50]. However, the investigation of potential structural and functional alterations in the BBB in neuropsychiatric diseases such as SZ, MDD, or ASD is still ongoing, probably due to varying degrees of inflammatory processes among the affected patient clusters (but not all) [51][52][51,52].
Studies have predominantly focused on alterations in the structure and function of proteins forming endothelial TJs, such as claudin-5, in neuropsychiatric disorders [53][54][53,54]. Additionally, other changes have been identified in preclinical and clinical studies using biomarkers, postmortem tissue, and neuroimaging [55]. Preclinical data are massive, and this resviearchw summarizes the principal clinical evidence:
-
Changes in the expression and function of P-glycoprotein [67] and other transcellular transporters (transferrin receptor (TfR), glucose transporter-1 (GLUT-1), insulin receptor (IR), and low-density lipoprotein receptor (LDLr)), which may impact the access of neuropsychiatric drugs and other promising pharmacological tools (e.g., nanoparticles, viral vectors) to the brain parenchyma through the BBB, as proposed for the treatment of neurological/neurodegenerative diseases [68][69][68,69].
Furthermore, the breakdown of the glymphatic system, mainly described in neurodegenerative diseases, is currently under investigation in psychiatric research due to its relevance to neuroinflammation and BBB permeability [70].
1.3. The Intestinal Barrier in Psychiatric Diseases
1.3.1. The Intestinal Barrier in Health and Disease
Besides its digestive and absorptive roles, the gastrointestinal tract exerts an effective but dynamic barrier function between the mucosal immune system and the vast array of microbial and alimentary antigens present in the intestinal lumen. Central to this barrier is the intestinal epithelial cells (IECs), which play vital roles both in generating immune tolerance and orchestrating effective innate and adaptive immune responses. The intestinal barrier encompasses not only IECs but also a variety of non-cellular elements including mucin, antimicrobial peptides, secretory immunoglobulin A (sIgA), and intercellular junction molecules between adjacent IECs [71][72][71,72].
Inflammatory cytokines, such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), can influence the anatomy and function of intercellular junctions [73], with a subsequent increased barrier permeability. This inflammation-associated dysfunction of the intestinal barrier can result in the translocation of commensal microorganisms and microbial products to the lamina propria, lymphatic vessels, and portal venous system. This translocation may trigger and perpetuate local and even systemic inflammatory processes. As a matter of fact, increased intestinal permeability is an early pathophysiological event in humans [74] and experimental [75] inflammatory bowel disease (IBD).
Additionally, experimental models of physical and psychological stress have demonstrated disruption of intestinal tight junctions, assuming their direct responsibility on the increased intestinal permeability observed in stressed laboratory animals [76]. In this sense, stressful events could trigger or modify the clinical course of IBD [77]. Moreover, the prevalence of psychiatric symptoms in IBD patients is substantial, with anxiety estimated to affect one-third and depression to one-quarter, especially those with active disease [78]
1.3.2. Intestinal Barrier Dysfunction in Psychiatric Diseases
Despite considerable efforts dedicated to studying the relationship between inflammatory balance and psychiatric pathology, there remain significant gaps in our understanding. Firstly, genetic vulnerability itself is insufficient for the onset of these diseases, suggesting the relevance of epigenetic changes induced by environmental factors, including inflammation. Secondly, the inflammatory response is nonspecific; several stimuli can trigger similar alterations in different psychiatric diseases, with slight nuances depending on whether it occurs in an early or chronic stage [79]. Thirdly, extensive longitudinal studies have failed to elucidate whether the activation of the immune system and the inflammatory response are the cause or consequence of these mental illnesses. Lastly, adjuvant anti-inflammatory therapies lead to moderate and non-stable symptom improvement, as inflammation is a homeostatic mechanism whose blockade could have counterproductive effects due to shared intra and intercellular pathways between pro- and anti-inflammatory mechanisms [80][81][80,81].
The origin of neuroimmune alterations in psychiatric diseases remains elusive. Epidemiological evidence suggests that peripheral immune alterations caused by circulating endotoxins through leaky internal barriers, including the intestinal barrier, could initiate an immune/inflammatory cascade in the brain, leading to structural or functional brain damage. This low-grade inflammation status, not an evident infection, seems a common factor in chronic diseases such as MDD and SZ [82]. At least in the initial years of the disease, stress is a prominent driving factor associated with inflammation in these pathologies. Other contributing factors include smoking, poor hygiene, diet, and metabolic changes induced by the disease or medications. However, another immune system mediator might play an important role in driving this inflammatory phenotype in psychiatric disorders: endotoxins.
Leaky gut and subsequent bacterial translocation have been identified clinically in MDD and SZ [83][84][83,84]. Recent research described different bacterial genera in fecal samples from MDD patients [85]. In SZ, leaky gut was significantly associated with a negative phenotype [86]. Stress-based experimental models of MD and SZ reported increased colony-forming units (CFUs) in tissues adjacent to the colon and elevated levels of LPS and LBP in plasma [87]. Structural and functional studies demonstrated that stress induces disruption of the intestinal barrier [39][88][39,88], and LPS from E Coli was observed to interact with innate immune receptors, such as Toll-like receptors (TLRs) expressed in brain areas [89]. Pharmacological and genetic studies using knockout mice indicate a role for endotoxins in depressive-like behaviors, identifying Gram-negative, Gram-positive, and anaerobic genera associated with bacterial translocation [39][87][39,87].
Several mechanisms have been proposed for the contribution of bacterial translocation to neuroimmune alterations in the CNS [90]. Peripheral proinflammatory cytokines associated with leaky gut may communicate with the brain via (a) the neural pathway, involving systemic cytokines directly activating primary afferent nerves (mainly the vagus); (b) the humoral pathway, through the choroid plexus and circumventricular organs (CVOs), which lack an intact BBB, allowing circulating cytokines to enter into the cerebral parenchyma through volume diffusion and elicit downstream signaling events important in altering brain function; (c) the cellular pathway, where systemic proinflammatory cytokines activate endothelial cells, leading to the activation of adjacent perivascular macrophages, which in turn activate microglia [91]. Additionally, LPS or other bacteria components from the gut can gain access to the circulation and directly invade the brain through a more permeable BBB or signal through leptomeningeal cells. Those cells express TLRs for LPS, and the released proinflammatory cytokines activate microglia, evoking neuroinflammation.
2. The Sphingosine-1-Phosphate (S1P) Metabolism and Signaling
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid that plays a crucial role in the immune system. It is widely expressed throughout the body and is involved in immune activation and regulation of cellular trafficking [92]. In addition to its immunological functions, S1P also influences other essential cellular processes, including barrier integrity, angiogenesis, and proliferation, through its synthesis in platelets, erythrocytes, vascular endothelial cells, and hepatocytes [93].
Many of the S1P actions are mediated through “inside-out signalling” wherein it acts as a ligand for a group of cell surface receptors belonging to the G-protein-coupled receptors (GPCRs) superfamily. S1P is transported to the extracellular milieu by specific proteins, such as sphingolipid receptor or spinster 2 (Spns2) [94], ATP-Binding cassette (ABC) transporters A1 (ABCA1) and C1 (ABCC1) [95][96][95,96], and major facilitator superfamily transporter 2b (Mfsd2b) [97]. These mechanisms allow S1P to exert its diverse effects on cellular responses and physiological processes throughout the body.
2.1. S1P Metabolism
Numerous studies have evinced the significance of phospholipids and their metabolites in various pathological conditions, including cardiovascular disorders, oncogenesis, and inflammatory diseases such as multiple sclerosis (MS) and IBD [97][98][99][100][97,98,99,100]. Among lipids, sphingolipids hold particular importance, characterized by the presence of sphingosine as their common base, which can be synthesized de novo or derived from a complex lipid hydrolysis pathway [101]. Ceramide (CER) plays a pivotal role in sphingolipid metabolism as the precursor of sphingosine, catalyzed by ceramidases [99]. CER and S1P exhibit opposing functions and the establishment of a CER/S1P rheostat is crucial for maintaining S1P gradients necessary for immune cell migration processes [102]. In humans, five ceramidases have been identified: acid ceramidase, neutral ceramidase, and alkaline ceramidase 1, 2, and 3, encoded by five different genes (ASAH1, ASAH2, ACER1, ACER2, and ACER3) [103]. Lastly, sphingosine kinases (Sphks) phosphorylate sphingosine to form S1P [104].
Of note, S1P synthesis is a complex mechanism characterized by its compartmentalization in different subcellar spaces due to its hydrophobic nature, which limits its metabolism to local enzymes. Consequently, S1P can exhibit diverse signaling properties depending on its cellular location [105]. The specific localization of S1P synthesis within distinct subcellular compartments gives rise to a complex network of vesicular and active protein mechanisms that regulate the transport of S1P across these compartments. As S1P is synthesized and localized in specific organelles, such as the endoplasmic reticulum (ER) and Golgi apparatus, it undergoes specific enzymatic modifications and interactions with other molecules before being transported to its intended target sites. These modifications and interactions can further regulate the signaling properties of S1P and contribute to its diverse cellular effects. Notably, in various tissues, intracellular S1P concentrations are maintained at low levels through rapid degradation facilitated by the S1P lyase (SGPL1) enzyme present in the ER. However, in contrast to this degradation pathway, S1P can also be transported out of the cell through specific transporters, enabling it to exert effects outside the cell [106].
2.1.1. S1P Synthesis Enzymes
To date, two distinct Sphk isoforms have been identified: sphingosine kinase 1 (Sphk1) and 2 (Sphk2) [107][108][107,108]. Although the activity of both isoforms overlaps to some extent, they differ in substrate specificity, temporal expression patterns during development, and subcellular localization, suggesting their involvement in different cellular processes [109]. Both Sphk1 and Sphk2 are necessary for maintaining physiological levels of S1P, but they play essential yet antagonistic roles.
Under proinflammatory stimuli, Sphk1 translocates to the plasma membrane to generate S1P as an intracellular messenger [105]. Notably, Sphk1 stimulates cell growth and survival while suppressing apoptosis [110], and it is a key factor in maintaining cellular homeostasis under stress conditions by interacting with protein kinase R (PKR) [111].
Sphk1 is regulated “inside-out” by a complex array of post-transcriptional, epigenetics, and post-translacional mechanisms that ultimately generate a pool of S1P [112]. Its primary location is the cytosol, from where it translocates to the cell membrane through a process mediated by the Ca2+-myristoil switch protein calcium and integrin-binding protein. Once there, its activation takes place via phosphorylation of the Ser 225 by the action of the extracellular signal-regulated kinase (ERK) [113]. The activation of Sphk1 is necessary for its pro-proliferative and pro-survival signaling [114] and begins with the binding of anionic lipids such as phosphatidylserine (PS), phosphatidic acid (PA), and phosphatidylinositols (PIs) [115][116][115,116], as well as growth factors and cytokines [117][118][117,118].
In contrast to Sphk1, the functions of Sphk2 depend on its subcellular localization and cell type. Sphk2 promotes apoptosis in the ER, regulates mitochondrial respiration in mitochondria, modulates gene expression and telomere integrity in the nucleus, and is present in the plasma membrane in cancer [119].
2.1.2. S1P Degradation Enzymes
The degradation of S1P is influenced by the cellular localization of specific enzymes involved in the process. S1P degradation occurs irreversibly due to the activity of SGPL1 and reversibly by two S1P phosphatases, SGPP1 and SGPP2 [120]. SGPL1 is located on the ER membrane, specifically facing the cytosolic side, and acts as an important regulator of S1P levels. This enzyme catalyzes the breakdown of S1P into hexadecenal and phosphoethanolamine, effectively reducing the intracellular concentration of S1P [121]. Previous studies have shown that loss-of-function mutations in SPGL1 result in the pathological accumulation of sphingolipid intermediates and lead to apoptosis induction [122].
On the other hand, SGPP1 is located on the ER membrane, facing the cytosolic side, while SGPP2 is located on the ER membrane, facing the luminal side [123][124][123,124]. Thereby, SGPP1 and SGPP2 can modulate S1P levels by catalyzing the dephosphorylation of S1P, converting it into sphingosine. These isoenzymes exhibit high specificity for sphingoid base phosphates, although they are expressed in different tissues [125]. The phosphorylation-dephosphorylation process allows for the dynamic regulation of S1P/CER and its downstream signaling.
Additionally, sphingosine can be converted back to CER by adding a fatty acid, catalyzed by ceramide synthase, constituting the sphingolipid recycling pathway [126]. The sphingolipid recycling pathway helps maintain the balance of sphingolipid levels and ensures the availability of ceramide for various cellular processes, including lipid signaling, membrane maintenance, and apoptosis regulation [127].
2.2. S1P Signaling
Although sphingolipids are well-known components of cell membranes, they also participate in both inter- and intracellular signaling. S1P, classified as an amphipathic lysophospholipid, possesses a polar head and a hydrophobic chain, allowing it to be released from the plasma membrane and act as an intracellular mediator and a ligand for S1P receptors (S1PR). Five isoforms have been described (S1PR1-5), belonging to the family of G-protein-coupled receptors (GPCR) coupled to αio, αq, or α12/13 proteins [128][129][130][128,129,130]. Consequently, critical signaling molecules such as phospholipase C (PLC), ERK, phosphoinositide 3-kinase (PI3K), and protein kinase B (Akt) are activated. Akt phosphorylates the third intracellular loop of S1PR1-2, leading to the stimulation of Rac, a member of the Rho family of GTPases [131]. These intracellular signaling events ultimately regulate processes such as angiogenesis, immunity, directed cell migration, proadhesion, and vascular permeability regulation during inflammatory responses in the endothelium, all of which are closely related to the scope of this review.
Numerous studies have demonstrated the crucial regulatory role of S1P in immune responses [93], where its primary function is to orchestrate the dynamic trafficking of lymphocytes and other immune cells, facilitating their migration into lymphoid tissue and subsequent egress to the blood [132][133][132,133]. In the CNS, S1P influences microglial and astrocytic activity and promotes vascular integrity. Furthermore, it is involved in the production of cytokines and chemokines through the indirect activation of TLR-4, contributing to the maintenance of cerebral homeostasis. The participation of S1P has also been demonstrated in inflammatory diseases including cancer and diabetes, in pathophysiological processes such as atherosclerosis and osteoporosis, and even in chronic diseases, disorders, and autoimmune diseases [134][135][136][134,135,136].
The knowledge about the functions of S1PRs is mainly based on the use (even at the clinical level) of S1PR modulators, such as fingolimod, ozanimod, siponimod, and others [137]. The mechanisms of action of these compounds may be complex, probably acting through multiple S1PRs and different intracellular signaling pathways [137][138][137,138]. Their classical mode of action is functional antagonism, based on internalization and subsequent degradation of S1PRs from the lymphocytes cell surface, preventing lymphocyte trafficking between the lymph node, blood, and CNS [139]. Furthermore, these compounds can cross the BBB and are capable of modulating the expression and signaling of S1PRs expressed on endothelial cells and brain parenchymal cellular populations [140].
S1PR1-5 are expressed by various subtypes of innate immune cells. While S1PR1 is expressed in most of these cells, S1PR2 is predominantly found in macrophages, eosinophils, mast cells, and monocytes [141]. Additionally, S1PR3 and S1PR4 are also expressed in neutrophils and dendritic cells [142]. Lastly, S1PR5 is present in circulating monocytes and NK cells [143]. The detailed implications of S1PRs in the immune response and barrier function will be further elucidated in the next section.
Figure 1 illustrates the metabolism and signaling of S1P.
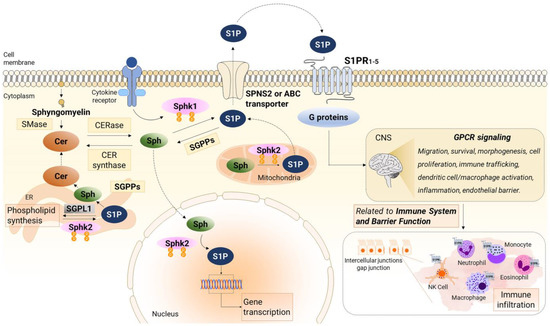
Figure 1. Sphingosine-1-phosphate (S1P) signaling and metabolism. Sphingosine kinase 1 (Sphk1) primarily resides in the cytosol and gets activated by cytokines, leading to the phosphorylation of Sphingosine (Sph) to generate S1P at the plasma membrane. Additionally, Sphingosine kinase 2 (Sphk2) phosphorylates Sph to produce S1P at various intracellular locations, such as the endoplasmic reticulum (ER), mitochondria, and nucleus. Once synthesized, S1P is exported from cells through a S1P transporter (SPNS2 protein or ABC transporter) and subsequently binds to specific S1P receptors (S1PRs). These receptors trigger downstream signaling pathways in an autocrine or paracrine manner, known as inside-out signaling. Within the ER, S1P is subject to degradation by S1P lyase or recycled for the synthesis of ceramide (CER) and complex sphingolipids. Moreover, S1P can also be dephosphorylated by phosphatase 1 (SGPP1) and 2 (SGPP2), both located in the ER, to form Sph, which is also reused for CER synthesis. The activation of S1PR1-5 initiates G-protein mediated signaling pathways that govern various cellular processes, including migration, survival, morphogenesis, cell proliferation, immune trafficking, dendritic cell/macrophage activation, inflammation, and endothelial barrier regulation associated with the immune system, and barrier function. The figure was prepared using the Motifolio Illustration Toolkits (Motifolio Inc., Ellicott City, MD, USA). SMase, sphingomyelinase; CERase, ceramidase; CER synthase, ceramide synthase.
3. S1P and the Immune System
As previously underscored, S1P signaling is essential in immune responses orchestrating the egress of lymphocytes from lymphoid tissues to blood. In the late 1990s, the new molecule FTY720 showed potent immunosuppressant activity leading to lymphopenia in rats [144] through a mechanism later identified as S1P-dependent [145], with S1PR1 playing a leading role [146]. These discoveries led to the emergence of FTY720, named fingolimod, as the first S1P-based drug approved for treating MS [100]. The proposed mechanism of action was, briefly, the retention of central memory T cells in lymph nodes by CC-chemokine receptor 7 (CCR7) after the aberrant internalization of S1PR1 caused by fingolimod. S1P levels are high in blood and lymph, helping immune cells to reach the vasculature and stabilizing the vessels. Fine-tuned gradients of S1P are critical for the exit and entrance of immune cells (not only T cells, but also B cells, NK cells mainly through S1PR5, and others) from primary and secondary lymphoid organs, and in nonlymphoid tissues. The precise mechanisms governing this traffic are not fully understood and the available evidence has been exhaustively reviewed both under physiological conditions [147] and after immune activation [148].
The expression patterns of S1PRs have emerged as key regulators in the inflammatory response. S1PR1 is almost ubiquitous being expressed in various cell types, including peripheral immune cells, endothelial cells, astrocytes, microglia, neurons, and to a lesser extent, oligodendrocytes [149]. Activation of S1PR1 on myeloid cells promotes neuroinflammation [150]. For its part, S1PR2 is widely expressed in various immune cells, including T cells, B cells, dendritic cells, macrophages, and natural killer (NK) cells, indicating its involvement in modulating immune responses and inflammatory processes. While S1PR2 predominates in proinflammatory cells, S1PR1 is also upregulated during the resolution phase of inflammation, facilitating macrophage migration from the inflammatory site and promoting the resolution process [151]. Both S1PR1 and S1PR2 signaling seem crucial for establishing and maintaining endothelial barrier function [152][153][152,153]. However, controversial data point in the opposite direction under specific circumstances, as discussed later.
S1PR3 is expressed in various cells and tissues, including endothelial cells, smooth muscle cells, neurons, glial cells, and cells of the immune system, such as T lymphocytes and dendritic cells. Under proinflammatory conditions, it can promote the migration of mature dendritic cells and mediate the chemotaxis of macrophages, neutrophils, and monocytes, driving leukocyte movement and recruitment to the site of inflammation [154]. Additionally, S1PR3 induces bactericidal action in macrophages through the production of reactive oxygen species (ROS), thereby enhancing the immune response [155].
S1PR4 is specifically expressed in lymphoid tissues and immune cells, including lymphocytes, dendritic cells, and macrophages. Its sole expression in these cell types suggests its relevance in immune responses and inflammatory processes, as it has been associated with cancer, autoimmune diseases, and IBD [92]. However, there is limited and controversial information about the mechanisms controlled by S1PR4. S1PR4 may participate in the release of proinflammatory cytokines in activated macrophages and mediate the activation and maturation of dendritic cells [156], but also its absence exacerbates M1 polarization and pulmonary inflammation [157].
S1PR5 is expressed in NK cells and patrolling monocytes. Similarly to S1PR4 and despite being one of the ozanimod targets, S1PR5 has been less studied. Available evidence points to its role in regulating NK cell migration, guiding their exit from the bone marrow and facilitating their circulation in the bloodstream [158], as well as in the egress of monocytes from the bone marrow and the inhibition of phagocytosis [159].
Beyond its peripheral participation in the immune cell migration toward the vasculature, S1P also regulates inflammation and immune events in the CNS. Most cell types in the CNS (neurons, microglia, astrocytes, oligodendrocytes, and BBB cells) express S1P receptors, and different S1P-based drugs have shown direct effects on them [160][161][160,161]. S1P, a lipid that easily accesses the CNS through the BBB, is the most enriched lipid in the CNS and plays an indubitable role in development. However, the neurotoxic/neuroprotective actions of S1P are still under debate, especially under disease conditions [162]. Some of the identified S1P pathways in the CNS are related to immune modulation, particularly, neuroinflammation. Glial activation is one of the main features of neuroinflammation and S1P may drive it. S1P accumulation has been shown to activate microglia in neural SPGL1 ablated mice through S1PR2 [163], in the microenvironment of degenerated intervertebral discs [164], and in glioma progression [165]. Both fingolimod and siponimods boost the expression of anti-inflammatory phenotypes and genes in microglia [166][167][166,167] and astrocytes [168]. Overexpression of S1PR1 is present in reactive astrocytes after fingolimod discontinuation [169] whereas this drug inhibits astrogliosis and its associated neuroinflammation in mice [170]. S1PRs are also involved in CXCL1 release from astrocytes [171] and CXCL5 from astrocytes and microglia after TLR4 stimulation [172]. All S1PR1-5 receptors, except S1PR4, have demonstrated participation in the activation of microglia and/or astrocytes, leading to neuroinflammation [173]. Nevertheless, their signaling is complex, as previously highlighted, and there are still controversial data that fuel an active debate about their exact neuroimmune mechanisms.
Beyond receptors, different pro/anti-inflammatory effects have been associated with metabolic S1P enzymes. Sphk1 overexpression plays a crucial role in the development of inflammatory and immune-related diseases, such as inflammatory bowel disease, Alzheimer’s disease, or hypertension. As a result, the antagonization of Sphk1 by Sphk1 or Sphk1/2 inhibitors is under investigation as a novel therapeutical alternative [174]. On the other hand, Sphk2 produces its own pool of S1P depending on its subcellular localization, which has been implicated in protection against ischemic injury, macrophage polarization, or regulation of cytokine expression [115]. However, there is scarce and controversial information on CNS diseases. Although the exact mechanisms behind the cytosolic/nuclear shift of Sphk2 remain elusive, a study suggests a neuroprotective activity of cytosolic Sphk2 in Alzheimer’s disease, as there is an inverse correlation between its cytosolic expression and amyloid deposits in the frontal cortex and hippocampus of AD patients, coinciding with translocation to the nucleus [114].
Consistent with the previously discussed functions of S1P, SGPL1 activity has been mostly associated with beneficial effects due to its anti-inflammatory properties [163][175][176][177][163,175,176,177] but it is also linked to deleterious effects on barrier function [178]. This ambivalent role depends on the cell type [179]. Similarly, the expression pattern of SGPP1 and SGPP2 in specific cell types may contribute differentially to the inflammatory context. SGPP2 activity is primarily upregulated during inflammation in many cells, such as endothelial cells and neutrophils [124]. In IBD models, SGPP2−/− mice showed less severe dextran sodium sulfate (DSS)-induced colitis together with the suppression of inflammation and intestinal cell apoptosis, leading to a healthier mucosal barrier. On the other hand, SGPP1 deletion implies a higher proinflammatory response after DSS-induced colitis [180].
The significance of S1P in bridging the activity of the innate and adaptive immune systems, coupled with its modulation of barrier functions discussed below, renders it a compelling target for controlling inflammation and the immune response in pathologies associated with immune dysfunction and barrier permeability, such as psychiatric diseases and IBD. Figure 2 presents an integrated view of the involvement of S1P signaling and metabolism molecules in these processes.
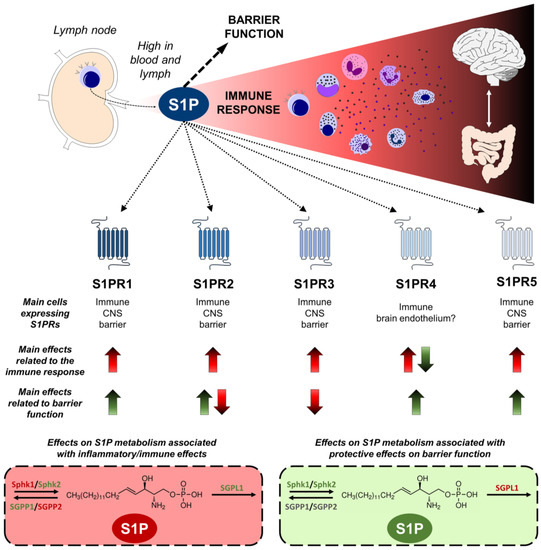
Figure 2. Sphingosine-1-phosphate (S1P) modulates the immune response and barrier function. S1P is a pivotal modulator of immune responses and barrier integrity. With elevated concentrations in the lymph and blood, S1P orchestrates immune cell trafficking from lymph nodes and contributes to inflammatory processes in several organs, including the central nervous system (CNS) and the gut, which are both relevant to psychiatric diseases. Five receptors (S1PR1-5) displaying distinct expression pattern in immune, CNS, and barrier-related cells, transduce the S1P signaling. Up arrows indicate an increase in the immune response or barrier function, and down arrows a decrease. Effects related to harmful situations are shown in red, and protective are in green. S1PR1, 2, 3, and 5 elicit inflammation and immune activation, while S1PR4 has shown ambivalent effects. The activity of S1PR1, 4, and 5 promotes a healthy barrier function, whereas S1PR3 exerts opposing effects, and S1PR2 yields controversial results. Similarly, S1P metabolism associated with inflammation includes the harmful activity of sphingosine kinase 1 (Sphk1) and sphingosine phosphatase 2 (SGPP2), and the suppression role of sphingosine kinase 2 (Sphk2), sphingosine phosphatase 1 (SGPP1), and S1P lyase 1 (SGPL1). Regarding protective effects on barriers, Sphk1 and 2 activation promote a healthier barrier, while SGPL1 is related to barrier dysfunction. The figure was prepared using the Motifolio Illustration Toolkits (Motifolio Inc., Ellicott City, MD, USA).