Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Hailong Chen.
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS), triggered by various pathogenic factors inside and outside the lungs, leads to diffuse lung injury and can result in respiratory failure and death, which are typical clinical critical emergencies. Severe acute pancreatitis (SAP), which has a poor clinical prognosis, is one of the most common diseases that induces ARDS. When SAP causes the body to produce a storm of inflammatory factors and even causes sepsis, clinicians will face a two-way choice between anti-inflammatory and anti-infection objectives while considering the damaged intestinal barrier and respiratory failure, which undoubtedly increases the difficulty of the diagnosis and treatment of SAP-ALI/ARDS.
- glucocorticoid
- severe acute pancreatitis
- acute lung injury
- acute respiratory distress syndrome
1. Introduction
Glucocorticoids (GCs) are “stress hormones” that affect the metabolic processes of almost every cell type. Under relaxed conditions, healthy individuals secrete about 20 mg GC/day but, under stress, secretion can reach 300 mg/day [1]. The circadian rhythm also regulates GCs production, which reaches its highest level in the blood during the early morning and drops to its lowest point at nightfall. This secretion pattern helps to regulate chemokine and inflammatory cytokine production and to moderate metabolism [2]. Each cell type has a distinct response to GCs and changes in GC structure and concentration affect the human body in different ways [3]. Meanwhile, GC function is dependent on its specific molecular structure, and GCs’ clinical efficacy is most dependent on the degree and duration of GC exposure to the GC receptor (GR). Maximal GRα saturation induced by different drug doses, start times, and mode and duration of administration ensures that GC efficacy is optimized [4]. In a randomized controlled trial of 17 ICU patients with moderate-to-severe ARDS, the administration of 20 mg/day intravenous GCs for 5 days followed by 10 mg/day from day 6 onwards downregulated inflammation, increased tissue repair, and elevated the alveolar-capillary membrane permeability index, reducing mechanical ventilation duration and overall mortality [5]. In recent decades, GCs have been increasingly used to treat several conditions, including various autoimmune diseases, rheumatoid arthritis, acute lung injury/acute respiratory distress syndrome (ALI/ARDS), sepsis, allergic skin diseases, and the suppression of transplant graft rejection [6]. GCs have also been shown to have a therapeutic effect against coronavirus disease 2019 (COVID-19). In a 2020 study of 173 patients with severe COVID-19 pneumonia from 14 distinct respiratory high-dependency units (RHDUs), early and extended administration of low-dose methylprednisolone (MP), a synthetic GC, improved the systemic inflammatory response, increased oxygenation markers, and reduced invasive mechanical ventilation and mortality rates [7].
Severe acute pancreatitis (SAP), which mediates the secretion of several inflammatory mediators and leads to ALI/ARDS, is a common clinical disease. ALI is the most important cause of the early death of SAP patients. Xuan et al. [8] surveyed 45 hospital ICUs and found that, while routine GC use remains controversial, most physicians still chose a short-course and low-dose GC treatment early in the disease process. However, several reports of adverse side effects have been recorded. GCs are often effective at improving clinical symptoms but rarely cure the disease. Thus, before prescribing GCs, it is important to define the corresponding parameters needed to decide whether to continue treatment [9].
2. GCs Mechanism in SAP-SIRS-ALI Treatment
22.1. Core Pathology of SAP-SIRS-ALI
2.1. Core Pathology of SAP-SIRS-ALI
Acute pancreatitis (AP) is a potential complication of abdominal surgery. Serious AP can lead to ALI/ARDS and is a common reason for AP-associated mortality. Currently, no treatment exists to slow progression. As a result of population growth, environmental changes (including changes in dietary patterns and increasing obesity and smoking rates), and improvements in diagnostic techniques, the annual global incidence of AP is increasing. In 2019, there were 34 cases of AP (95% CI: 23–49) per 100,000 in the general population and 1.16 deaths (95% CI: 0.85–1.58) per 100,000 in the general population [52][10]. This common and complex disease is also putting a major burden on national health care costs. AP-related admission costs in the United States were USD 2.2 billion in 2003 (95% CI: 2.0–2.3 billion), with a mean cost per hospital day of USD 1670 (95% CI: 1620–1720), and an average cost per hospital stay of >USD 10,000. These findings underscore the importance of identifying cost-effective treatment strategies and illustrate that the early identification and prevention of disease progression can significantly reduce hospital costs [53][11]. The mechanisms involved in the pathological process of SAP causing ALI/ARDS have been outlined by ourthe research team in a previous review. Damage to acinar cells is triggered by abnormal activation of pancreatic trypsin induced by various etiologies including glandular pathological changes such as calcium overload of alveolar cells and the disruption of pancreatic microcirculation. This promotes the destruction of mechanical, chemical, biological, and immune barriers in the intestine. Accompanied by inflammatory cell necrosis and the high production of inflammatory mediators such as IL-1β and TNF-α, this inflammatory storm increases local inflammation to SIRS, indicating that AP has begun the early stage of multi-organ dysfunction syndrome (MODS). After extensive damage to alveolar epithelial cells, capillary endothelial cells, and the blood–brain barrier caused by elevated inflammatory cell numbers and cytokine production, ALI/ARDS begins to develop [54][12]. The 1992 American College of Chest Physicians/Society of Critical Care Medicine (SCCM) Consensus Conference Committee defines this as an uncontrolled host response to infectious or non-infectious injury [55,56][13][14]. Excessive cytokine production disrupts the balance of pro- and anti-inflammatory cytokines and signals the development of SIRS. The inflammatory mediators involved in this pathological process include tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10), intercellular adhesion molecule-1 (ICAM-1), platelet-activating factor (PAF), CD40L, complement component C5a, substance P, hydrogen sulfide (H2S), and chemokines [57,58,59,60,61][15][16][17][18][19]. SIRS is a double-edged sword for the body. While fighting pathogens or the body’s own necrotic tissue, SIRS can have many harmful effects including altered immune status, organ dysfunction, and a disruption in the procoagulant/anticoagulant balance [62][20]. There are two mortality peaks associated with SAP (Figure 71). The initial peak occurs 1–2 weeks following the onset of symptoms. MODS, which is induced by SIRS, is the most common cause of death at this time point, accounting for 60–80% of all fatalities. The second peak occurs approximately 2 months after onset. Most of these deaths are caused by sepsis and other infections, and the length of SIRS is directly associated with the severity of SAP and its prognosis [63][21]. Hietaranta et al. [55][13] showed that the risk of systemic complications increases as the severity of SIRS rises.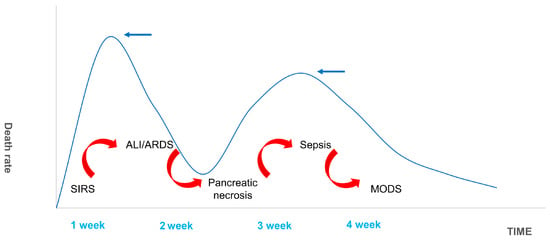
Figure 71. Death Spikes of SAP. The figure shows the distribution of deaths in SAP patients. The first peak of death occurred in the first week or so after SAP, and respiratory failure caused by ALI/ARDS complicated by SIRS was the most common. The second death peak was due to sepsis secondary to MODS caused by pancreatic necrosis infection. It is usually located in the third and fourth weeks.
3.2. Research Progress of GC in SAP-SIRS-ALI
2.2. Research Progress of GC in SAP-SIRS-ALI
GCs are the most used drug for the treatment of ALI/ARDS caused by SAP and can effectively reduce patient mortality. After binding to GR and moving into the nucleus, GCs play a significant role in promoting an anti-inflammatory response, reducing oxygen-free radical damage, and improving microcirculation [70][28]. Kimura et al. [71][29] found that, following adrenalectomy, AP rats were more sensitive to acinar cell apoptosis due to reduced endogenous cortisol production. These animals had greater pancreatic edema, higher amylase levels, more potent inflammatory responses, and higher rates of mortality. These effects were significantly reduced following exogenous GC administration [72][30]. These studies provide evidence that GCs reduce the onset of AP [73][31].References
- Kemppainen, R.J.; Behrend, E.N. Adrenal physiology. Vet. Clin. N. Am. Small Anim. Pract. 1997, 27, 173–186.
- Shimba, A.; Ikuta, K. Glucocorticoids Regulate Circadian Rhythm of Innate and Adaptive Immunity. Front. Immunol. 2020, 11, 2143.
- Wilcke, J.R.; Davis, L.E. Review of glucocorticoid pharmacology. Vet. Clin. N. Am. Small Anim. Pract. 1982, 12, 3–17.
- Meduri, G.U.; Annane, D.; Confalonieri, M.; Chrousos, G.P.; Rochwerg, B.; Busby, A.; Ruaro, B.; Meibohm, B. Pharmacological principles guiding prolonged glucocorticoid treatment in ARDS. Intensive Care Med. 2020, 46, 2284–2296.
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A.; et al. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276.
- Dubashynskaya, N.V.; Bokatyi, A.N.; Skorik, Y.A. Dexamethasone Conjugates: Synthetic Approaches and Medical Prospects. Biomedicines 2021, 9, 341.
- Salton, F.; Confalonieri, P.; Meduri, G.U.; Santus, P.; Harari, S.; Scala, R.; Lanini, S.; Vertui, V.; Oggionni, T.; Caminati, A.; et al. Prolonged Low-Dose Methylprednisolone in Patients with Severe COVID-19 Pneumonia. Open Forum Infect. Dis. 2020, 7, ofaa421.
- Xuan, N.; Zhang, X.; Hu, W.; Chen, G.; Wang, Y.; Zhang, S.; Cui, W.; Zhang, G. Effects of the working experience, educational background, professional titles, and hospital grades of intensive care unit doctors on clinical glucocorticoid use in acute respiratory distress syndrome. Medicine 2022, 101, e29021.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545.
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 175–184.
- Fagenholz, P.J.; Fernández-del Castillo, C.; Harris, N.S.; Pelletier, A.J.; Camargo, C.A., Jr. Direct medical costs of acute pancreatitis hospitalizations in the United States. Pancreas 2007, 35, 302–307.
- Ge, P.; Luo, Y.; Okoye, C.S.; Chen, H.; Liu, J.; Zhang, G.; Xu, C.; Chen, H. Intestinal barrier damage, systemic inflammatory response syndrome, and acute lung injury: A troublesome trio for acute pancreatitis. Biomed. Pharmacother 2020, 132, 110770.
- Hietaranta, A.; Kemppainen, E.; Puolakkainen, P.; Sainio, V.; Haapiainen, R.; Peuravuori, H.; Kivilaakso, E.; Nevalainen, T. Extracellular phospholipases A2 in relation to systemic inflammatory response syndrome (SIRS) and systemic complications in severe acute pancreatitis. Pancreas 1999, 18, 385–391.
- Rangel-Frausto, M.S.; Pittet, D.; Costigan, M.; Hwang, T.; Davis, C.S.; Wenzel, R.P. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA 1995, 273, 117–123.
- Bhatia, M. Novel therapeutic targets for acute pancreatitis and associated multiple organ dysfunction syndrome. Curr. Drug Targets-Inflamm. Allergy 2002, 1, 343–351.
- Bhatia, M. Inflammatory response on the pancreatic acinar cell injury. Scand. J. Surg. 2005, 94, 97–102.
- Bhatia, M.; Brady, M.; Shokuhi, S.; Christmas, S.; Neoptolemos, J.P.; Slavin, J. Inflammatory mediators in acute pancreatitis. J. Pathol. 2000, 190, 117–125.
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156.
- Bhatia, M.; Wong, F.L.; Cao, Y.; Lau, H.Y.; Huang, J.; Puneet, P.; Chevali, L. Pathophysiology of acute pancreatitis. Pancreatology 2005, 5, 132–144.
- Adib-Conquy, M.; Cavaillon, J.M. Compensatory anti-inflammatory response syndrome. Thromb. Haemost. 2009, 101, 36–47.
- Xiong, J.; Zhu, S.; Zhou, Y.; Wu, H.; Wang, C. Regulation of omega-3 fish oil emulsion on the SIRS during the initial stage of severe acute pancreatitis. J. Huazhong Univ. Sci. Technol. 2009, 29, 35–38.
- Ward, N.S.; Casserly, B.; Ayala, A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest Med. 2008, 29, 617–625.
- Bone, R.C. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit. Care Med. 1996, 24, 1125–1128.
- Gunjaca, I.; Zunic, J.; Gunjaca, M.; Kovac, Z. Circulating cytokine levels in acute pancreatitis-model of SIRS/CARS can help in the clinical assessment of disease severity. Inflammation 2012, 35, 758–763.
- Osuchowski, M.F.; Welch, K.; Siddiqui, J.; Remick, D.G. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 2006, 177, 1967–1974.
- Zhang, X.P.; Zhang, L.; Chen, L.J.; Cheng, Q.H.; Wang, J.M.; Cai, W.; Shen, H.P.; Cai, J. Influence of dexamethasone on inflammatory mediators and NF-kappaB expression in multiple organs of rats with severe acute pancreatitis. World J. Gastroenterol. 2007, 13, 548–556.
- Zhang, X.P.; Zhang, L.; Wang, Y.; Cheng, Q.H.; Wang, J.M.; Cai, W.; Shen, H.P.; Cai, J. Study of the protective effects of dexamethasone on multiple organ injury in rats with severe acute pancreatitis. Jop 2007, 8, 400–412.
- Zhao, Y.; Xiong, R.P.; Chen, X.; Li, P.; Ning, Y.L.; Yang, N.; Peng, Y.; Jiang, Y.L.; Zhou, Y.G. Hsp90 regulation affects the treatment of glucocorticoid for pancreatitis-induced lung injury. Mol. Cell. Biochem. 2018, 440, 189–197.
- Kimura, K.; Shimosegawa, T.; Sasano, H.; Abe, R.; Satoh, A.; Masamune, A.; Koizumi, M.; Nagura, H.; Toyota, T. Endogenous glucocorticoids decrease the acinar cell sensitivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology 1998, 114, 372–381.
- Abe, R.; Shimosegawa, T.; Kimura, K.; Abe, T.; Kashimura, J.; Koizumi, M.; Toyota, T. The role of endogenous glucocorticoids in rat experimental models of acute pancreatitis. Gastroenterology 1995, 109, 933–943.
- Muller, C.A.; Vogeser, M.; Belyaev, O.; Gloor, B.; Strobel, O.; Weyhe, D.; Werner, J.; Borgstrom, A.; Buchler, M.W.; Uhl, W. Role of endogenous glucocorticoid metabolism in human acute pancreatitis. Crit. Care Med. 2006, 34, 1060–1066.
More