Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Paola Venditti and Version 2 by Jason Zhu.
Aerobic organisms use molecular oxygen in several reactions, including those in which the oxidation of substrate molecules is coupled to oxygen reduction to produce large amounts of metabolic energy. The utilization of oxygen is associated with the production of ROS, which can damage biological macromolecules but also act as signaling molecules, regulating numerous cellular processes. Mitochondria are the cellular sites where most of the metabolic energy is produced and perform numerous physiological functions by acting as regulatory hubs of cellular metabolism.
- ROS production
- oxidative damage
- mitochondrial dynamics
1. Mitochondrial Fusion and Fission
Mitochondria are subject to continuous rearrangement and turnover that aim to preserve their functionality. Maintaining a healthy mitochondrial population is crucial to safeguarding cell function and the energy homeostasis of the whole body.
The rate at which the mitochondrial pool renews depends on the balance between mitochondrial biogenesis and fusion, fission, or degradation through mitophagy processes. The processes of biogenesis and mitophagy generate new mitochondria and eliminate them, respectively. Mitophagy is relevant both for the adaptation of the mitochondria’s content to the cell’s metabolic needs and the removal of damaged organelles. Furthermore, several data points suggest that mitochondrial fission followed by selective fusion is a prerequisite for the degradation of dysfunctional mitochondria via autophagy [1][87]. Alterations in the delicate balance of the processes involved in mitochondrial turnover (mitophagy, mitochondrial biogenesis, and mitochondrial dynamics) can play a crucial role in the aging process [2][88]. In nematodes, mitochondria gradually accumulate with age, and depletion of the master regulator of autophagy recapitulates the effects of aging on mitochondrial mass in young adult animals [3][89]. The impairment of mitophagy reduces the resistance to stress and activates a signaling pathway that, starting from the mitochondria, regulates genes involved in both mitochondrial biogenesis and mitophagy [3][89]. These data suggest the existence of a feedback loop capable of integrating metabolic signals to coordinate mitochondrial biogenesis and turnover. The uncoupling of these two processes during aging contributes to the excessive accumulation of damaged mitochondria and the decline of cellular function [3][89].
For the understanding of the molecular mechanisms underlying mitophagy, the discovery of the pathway mediated by phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-Parkin was crucial [4][5][6][90,91,92]. The protein PINK1 was identified in 2001 [7][93]. It is a serine/threonine kinase that possesses a mitochondrial targeting sequence at the N-terminal site and a signal that allows localization at the outer mitochondrial membrane (OMM) [8][94]. Parkin was discovered in 1998 and is a ubiquitin E3 ligase whose name is linked to its relevant role in the pathogenesis of juvenile parkinsonism [9][95].
Briefly, PINK1 is continuously degraded in a complex process that includes its importation into the mitochondrial matrix, where it undergoes cleavage by both mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protease (PARL). Then, PINK1 is translocated back to the cytosol and degraded via the N-end rule pathway [10][96].
When mitochondrial damage occurs, such as a fall in membrane potential or accumulation of misfolded proteins, the PINK1- and Parkin-dependent mitophagy pathways are activated [5][91]. PINK1 is stabilized at the OMM, where it undergoes autophosphorylation and then phosphorylates Parkin, activating Parkin ligase E3Ub activity and its recruitment to damaged mitochondria [1][87]. Activated Parkin adds ubiquitins to several mitochondrial substrates, including mitofusin 1 and mitofusin 2 (MFN1/2) [11][97], voltage-gated anion channel 1 (VDAC1) [12][98], PARIS [13][99], and Miro [14][100]. Polyubiquitination of mitochondrial proteins induces association with autophagy receptors, thereby resulting in the formation of the autophagosome [15][16][101,102], which subsequently fuses with lysosomes, thus favoring the degradation of mitochondria [17][103].
In summary, damaged mitochondria are identified and labeled by parkin-dependent ubiquitination of certain proteins and eliminated. However, there is also an alternative pathway for mitophagy initiation since PINK1 can directly recruit autophagy receptors independently of Parkin [17][103].
Moreover, mitophagy is tightly regulated by the processes of fusion and fission, also indicated as “mitochondrial dynamics” [18][104] (Figure 1). Fusion occurs when two adjoining mitochondria merge, forming a longer mitochondrion, while fission occurs when one mitochondrion separates into two [19][105].
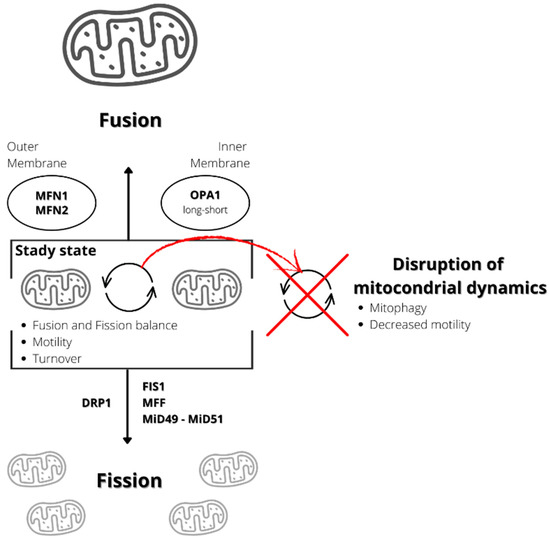
Figure 1. Schematization of mitochondrial dynamic. Mitochondrial fission produces two mitochondria from one mitochondrion, while fusion determines the production of one mitochondrion from two. Proteins involved in the fission processes are dynamin-related protein 1 (DRP1), fission 1 homolog protein (FIS1), mitochondrial fission factor (MFF), mitochondrial dynamics protein of 49 kDa (MiD49), and mitochondrial dynamics protein of 51 kDa/mitochondrial elongation factor 1 (MiD51/MIEF1). The proteins involved in the fusion process are mitofusins 1 and 2 (MFN1 and MFN2), involved in the outer membrane fusion, and OPA1 (long and short forms), involved in the intra-membrane fusion. Disruption in the fusion/fission balance can reduce mitochondrial motility, inducing mitophagy.
Fusion and fission processes are both regulated by specific proteins. The proteins involved in the process of fusion are three GTPases, two of which localize at the outer mitochondrial membrane, MFN1 and MFN2, and one located at the inner mitochondrial membrane and intermembrane space, optic atrophy gene 1 (OPA1) [20][106]. Fusion determines the mixing of the content of mitochondrial matrixes, intermembrane spaces, and the mtDNA molecules of two mitochondria. This process can be relevant to buffering, at least partially, defects and transient stresses. Indeed, it enables optimal mitochondrial function by diluting mutated mtDNA and salvaging damaged mitochondria by acquiring key components from healthy mitochondria [21][107].
The proteins involved in mitochondrial fission include a soluble cytosolic protein, the dynamin-related protein 1 (DRP1), a member of the dynamin family of GTPases, and mitochondria-bound proteins, including fission 1 homolog protein (FIS1), mitochondrial fission factor (MFF), mitochondrial dynamics protein of 49 kDa (MiD49), and mitochondrial dynamics protein of 51 kDa/mitochondrial elongation factor 1 (MiD51/MIEF1) [20][106]. Mitochondrial outer membrane contact sites with the endoplasmic reticulum that define the position where the constriction machinery assembles, and division occurs regulate the process [22][23][24][25][108,109,110,111]. DRP1 is not anchored to the membrane and is recruited from the cytoplasm to homo-oligomerize around the outer mitochondrial membrane to constrict it in a process that GTP binding and hydrolysis drive [26][112].
Following a fission event, the daughter mitochondrion may either maintain intact membrane potential or depolarize. If it depolarizes, it is unlikely to proceed to a subsequent fusion unless it repolarizes. After being depolarized and solitary for a few hours, the mitochondrion is removed by autophagy [27][113].
Mitochondrial dynamics regulate mitophagy and contribute to mitochondrial quality control. In mouse pancreatic β-cells, mitochondrial fission results in the identification of damaged mitochondria in the mitochondrial population and their subsequent removal by mitophagy [27][113]. Thus, defects in mitochondrial fusion or fission processes can cause defects in mitochondrial function and activity because mitochondrial dynamics involve multiple processes such as fission and fusion equilibrium, mitochondrial turnover, and motility. However, motility and turnover depend on fusion and fission [28][114]. Indeed, in neurons, defects in both fusion and fission lead to reduced mitochondrial motility and neuronal dysfunction [28][114]. In summary, mitochondrial dynamics impairment seems to be involved in several diseases [18][104].
Furthermore, unbalanced mitochondrial dynamics can alter mitophagy by preventing the elimination of damaged mitochondria. Alterations of this type are found in various age-related diseases such as sarcopenia [29][115], Alzheimer’s disease [30][116], obesity, and metabolic alterations [31][117].
Many proteins involved in mitochondrial fission dysregulate with aging. In old mice, the activity of DRP1 reduces, and this is associated with altered morphology of mitochondria in neurons, skeletal muscle, and oocytes [32][33][118,119]. Moreover, in the skeletal muscle of aged mice, there is an increased ratio between MFN2/DRP1 and longer intermyofibrillar mitochondria [34][120]. In human endothelial cells from old subjects, the downregulation of both DRP1 and FIS1 expression is associated with an elongated mitochondrial network [35][121]. Moreover, with aging, the expression of MFN2 is also downregulated, suggesting a link between reduced fusion and dysfunction in mitochondrial dynamics [29][115].
Several pieces of evidence obtained in many model organisms suggest that changes in the process of mitophagy can affect health and lifespan. It has been reported that the mitophagy rate in 21-month-old mice is not as high as that observed in 3-month-old mice in the dentate gyrus of the hippocampus [36][122]. Defects in mitophagy have been observed in the satellite cells of the skeletal muscles of aged humans and mice [37][123] and in the hearts of aged mice [38][124]. However, the finding of reduced expression of genes encoding proteins involved in mitophagy in the skeletal muscle of physically inactive but not active older women suggests that exercise while preserving muscle mass may protect mitophagy [39][125].
During aging, both dysfunctional mitochondrial dynamics and inefficient mitophagy can determine the accumulation of damaged and malfunctioning mitochondria in cells, which can impair cellular function (Figure 2). Indeed, it has been reported that the age-related ROS increase in sperm induces mitochondrial damage due to impaired mitophagy [40][126]. Furthermore, the reduced telomerase length could induce an activation of p53 that suppresses PGC-1α, impairing mitochondrial function and mitochondrial DNA content [41][127] (Figure 2).
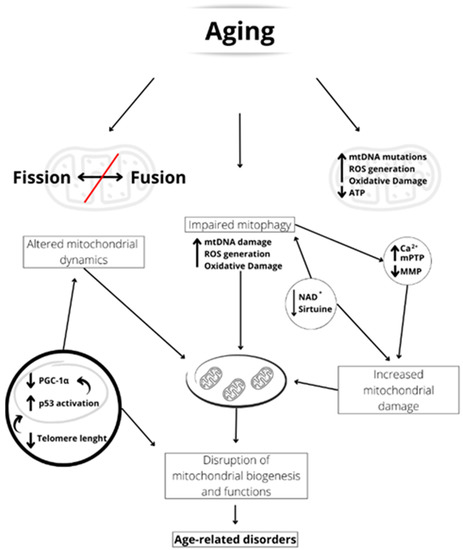
Figure 2. Schematic representation of the aging-linked mitochondrial dysfunctions. Aging can induce unbalanced fusion/fission dynamics, accumulation of mitochondrial damage, and impaired mitophagy. The shortened telomerase length activates p53, which reduces PGC-1α expression and, therefore, mitochondrial biogenesis. Furthermore, reduced NAD+ concentration and sirtuin activity led to impaired mitophagy. This may result in increased mitochondrial [Ca2+], mPTP formation, and inner mitochondrial membrane potential (MMP) reduction, leading to increased mitochondrial damage. All these factors induce an alteration of mitochondrial biogenesis and functionality, which are at the basis of age-related disorders.
2. Mitochondrial Biogenesis
The maintenance of efficient mitochondria is necessary for cellular homeostasis and is achieved through the continuous removal of defective mitochondria and the synthesis of new mitochondria (mitochondrial biogenesis) to maintain a mass of good-quality mitochondria to meet cellular energy needs [3][89].
The biogenesis of the mitochondria is a tightly regulated process that involves several transcription factors and the coordination between the mitochondrial and nuclear genomes in a complex and multistep process. The factors involved in the biogenesis process include the nuclear respiratory factors 1 and 2 (NRF1 and NRF2) and transcriptional coactivators, such as members of the peroxisome proliferator-activated receptor γ coactivator-1 family (PGC1α, PGC1β, and PGC1 related coactivator, PRC), of which PGC1α is considered the master regulator of mitochondrial biogenesis, estrogen-related receptors (ERRα, ERRβ, and ERRγ), and mitochondrial transcription factor A (TFAM) [42][128]. Transcription of the mtDNA starts with the activation, due to either phosphorylation or deacetylation, of PGC1α, followed by stimulation of NRF1, NRF2, and ERRα, with consequent increased expression of TFAM, which acts as the final effector of mtDNA transcription [43][129].
The translation of genes encoded by mtDNA into proteins requires the participation of translation factors encoded by nuclear DNA: the initiation factors 2 and 3 (mtIF2 and mtIF3), the elongation factors Tu, Ts, and G1 (mtEFTu, mtEFTs, and mtEFG1), the translational release factor1-like (mtRF1L), and the recycling factors (mtRRF1 and mtRRF2). The translational activator of cytochrome c oxidase 1 (TACO1) that binds to the mRNA regulates the levels of mitochondrial proteins [43][129].
NRF1 and NRF2 directly regulate the expression of nuclear-encoded mitochondrial genes through the production of preprotein [42][128]. Therefore, the proteins encoded by nDNA derive from preproteins synthesized within the cytosol and containing an amino terminal signal that targets them to the mitochondria. Protein precursors with a cleavable pre-sequence are imported by the pre-sequence translocase (TIM23 complex), while other precursors containing internal targeting signals are imported by the carrier translocase (TIM22 complex) [44][130]. TIM23 and TIM22 are both activated by the transmembrane electrochemical potential. Many small inner membrane proteins do not resemble canonical TIM23 or TIM22 substrates, and their import mechanism is unknown [44][130].
In summary, both NRF1 and NRF2 are implicated in the expression of mitochondrial respiratory chain proteins, proteins that play the role of promoting the import of preproteins or are involved in the biosynthesis of heme groups, and transcription factors of mtDNA, such as TFAM [42][45][128,131]. PGC1α is the master regulator of mitochondrial biogenesis by co-activating the expression of the transcription factors NRF1, NRF2, and ERRα [46][132].
Several experimental studies suggest that a reduction in mitochondrial biogenesis may be involved in the aging process. It has been found that there is a link between telomere damage and mitochondrial dysfunction [47][133]. Telomeres are structures possessing repetitive nucleotide sequences and specific binding proteins at the terminal region of the chromosomes of eukaryotes, forming nucleoprotein complexes that assure genomic stability [48][49][50][134,135,136]. In somatic cells, a progressive shortening of telomeres occurs with each division due to the lack of telomerase, an enzyme that adds nucleotides to telomeres. Progressive shortening eventually leads to replicative senescence [51][52][137,138], and, in humans, the length of the telomeres in the blood correlates with health and lifespan in 60-year-old or older subjects [53][139], even if it has been reported that lifespan is mainly related to the percentage of short telomeres rather than the average telomere length [54][140].
The master regulator of the correlation between telomere length and mitochondrial dysfunction is the nuclear transcription factor protein 53 (p53), which transactivates numerous target genes involved in the induction of cell cycle arrest and/or apoptosis [55][56][57][58][141,142,143,144], and PGC coactivators of transcription [47][133]. The shortening of the telomeres is associated with increased p53 and high levels of apoptosis [59][145]. Indeed, activated p53 promotes the arrest of the cell cycle, or apoptosis, to prevent the propagation of cells with severe DNA damage [60][146]. Activated p53 can interact with the promoters of PGC1A and PGC1B, determining the suppression of their expression. The reduction in PGC coactivators determines the reduction in mitochondrial biogenesis and functionality, thus leading to functional decline and aging in tissue stem cells and postmitotic tissues [47][133]. More importantly, forced expression of PGC1α or deletion of p53 in the context of telomere dysfunction restores mitochondrial respiration [47][133]. The mitochondrial decline occurring during physiological aging in wild-type mice can be partially reversed by telomerase activation [61][147], confirming the link between telomere length and mitochondrial efficiency.
Alterations of mitochondrial biogenesis with age also correlate with a reduction in nicotinamide adenine dinucleotide (NAD+) levels [62][148], which can compromise the activity of sirtuin deacetylases, of which NAD+ is a co-substrate (Figure 2) [63][149]. Seven sirtuin enzymes, from SIRT1 to SIRT7, have been identified in mammals, three of which, SIRT3, SIRT4, and SIRT5, are found in the mitochondrial matrix [63][149]. The dependence of sirtuins on NAD+ makes them dependent on the metabolic state of the cell and sensors of stress [63][149]. For example, it has been found that SIRT3 increases mitochondrial oxidative metabolism in response to nutrient stress and membrane depolarization [64][65][150,151] and that its loss decreases cardiac function and induces neurodegeneration [66][67][152,153].
Moreover, SIRT3 activates by deacetylation not only enzymes regulating mitochondrial metabolism, such as isocitrate dehydrogenase or complexes of the mitochondrial respiratory chain, but also enzymes involved in the cellular response to oxidative stress [68][154].
SIRT3 also contributes to preserving cardiac function in stressful conditions. Indeed, it has been found that the mitochondrial fusion protein OPA1 is post-translationally modified in the heart by hyperacetylation under pathological stress, which reduces its GTPase activity [69][155]. SIRT3 deacetylating OPA1 elevates OPA1 GTPase activity [69][155]. The activation of OPA1 by SIRT3 contributes to preserving the mitochondrial network, protecting the cardiomyocytes from doxorubicin-mediated cell death [69][155].
The involvement of SIRT3 in mitochondrial biogenesis is also suggested by the observation that silencing it in SW620 cancer cells induces decreased mitochondrial biogenesis and mitochondrial dysfunctions, as shown by reduced protein content of PGC1α and TFAM and reduced amounts of the complexes of the mitochondrial chain [70][156]. These data suggest that SIRT3 regulates mitochondrial homeostasis at several levels, including mitochondrial biogenesis and function.
Lastly, sirtuins can also modulate autophagy, the cellular-relevant process that allows the lysosomal-mediated elimination of damaged proteins and organelles, including mitochondria, endoplasmic reticulum, and peroxisomes. SIRT1 has been shown to be able to deacetylate p53, resulting in the reduction of apoptosis and the induction of autophagy [71][157]. Sirtuin-induced activation of autophagy can also reduce cellular senescence, as demonstrated by the effects of SIRT6 overexpression on human bronchial epithelial cells, in which senescence was induced by cigarette smoke extract treatment [72][158]. Furthermore, the senescence process is increased in both SIRT6 knockdown mice and mice with mutant SIRT6 without histone deacetylase activity [72][158]. Finally, it has been shown that miR-212-mediated negative regulation of SIRT1-dependent autophagy increases the senescence of cells [73][159]. In conclusion, it is possible that regulation of the sirtuins that play a role in the biogenesis of mitochondria [74][160] and the elimination of damaged mitochondria by autophagy [75][161] can modulate the function of mitochondria and play a protective role in age-associated diseases.