Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Adam C Midgley and Version 2 by Conner Chen.
At least three local events are needed to generate mature α-SMA-positive, fully differentiated myofibroblasts: (i) biologically active TGF-β1; (ii) extracellular stress, arising from the mechanical properties of the ECM (particularly collagen) and EDA–FN/integrin interactions; and (iii) the precursory production of phenotypic modulators (EDA–FN, HA) following activation. Increasing evidence strongly supports the role of inflammatory cell interactions in promoting myofibroblast development. Regardless of the origin, the resultant myofibroblast cells share the same properties and signalling cascade events that led to their formation.
- myofibroblast
- fibrosis
- wound healing
- Transforming growth factor
- mechanotransduction
1. The Canonical Pathway: TGF-β1/Smad
In classical cutaneous wound healing, tissue injury leads to TGF-β1 release from keratinocytes, macrophages and degranulating platelets [1][46]. In turn, TGF-β1 binds to and induces TGF-β receptor (TGFβR) I and II association on the fibroblast cell membrane, enabling TGFβRII phosphorylation of the TGFβRI kinase domain. In-turn, TGFβRI phosphorylates Smad2 and Smad3, which subsequently oligomerize with Smad4, forming trimeric protein complexes. These complexes are translocated to the nucleus, where they act as the transcription or co-transcription factors in the induction or repression of gene expression [2][47] (Figure 12A). Ultimately, TGF-β1/Smad pathway activation in fibroblasts leads to their differentiation into myofibroblasts [3][48]. TGF-β1 can also induce Smad-independent and co-receptor signalling pathways, such as mitogen-activated protein kinase (MAPK), p42/p44 extracellular signal regulated kinase (ERK1/2), Rho/Rho-associated protein kinase (ROCK), phosphatidylinositol-3-kinase (PI3K)/AKT, protein phosphatase 2A (PP2A), p38/c-Jun N-terminal kinase (JNK), protein kinase C (PKC), and tumour necrosis factor receptor associated factor (TRAF)-4/6 [4][5][6][7][8][49,50,51,52,53]. The tightly controlled TGF-β1/Smad-driven signalling events have been extensively researched within the context of myofibroblast differentiation and fibrosis, as highlighted in an eloquent review by Frangogiannis [9][54].
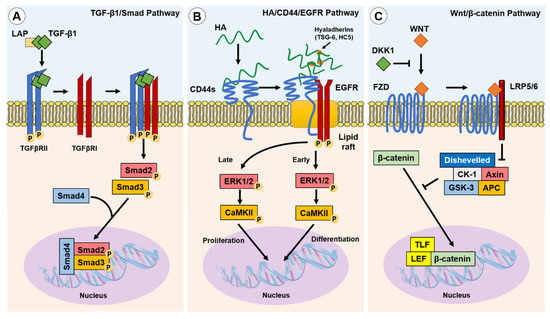
Figure 12. Canonical and non-canonical pathways implicated in fibroblast–myofibroblast differentiation. Three of the best described pathways involved in myofibroblast phenotypic acquisition are the TGF-β1/Smad; HA/CD44/EGFR; and Wnt/β-catenin pathways: (A) TGF-β1 is released from the LAP complex and binds to TGFβRII dimers, enabling association with and the activation of TGFβRI. Smad2 and Smad3 are subsequently phosphorylated and co-associate with Smad4 to initiate the transcription of pro-fibrotic genes; (B) TGF-β1 induced the upregulation of HAS2 synthesised linear HA and hyaladherin crosslinkers, such as TSG-6 and HC5, resulting in HA interactions with CD44s and the thickening of a crosslinked HA pericellular coat. HA interactions with CD44s induce clustering of CD44s within lipid rafts, wherein the co-receptor EGFR is activated and initiates biphasic signalling via ERK1/2 and CaMKII. Early signalling was suggested to promote differentiation-associated gene expression, whereas late signalling was suggested to promote proliferation-associated gene expression; and (C) the Wnt ligand binding to the transmembrane Frazzled receptor (FZD), in the absence of DKK1, which induces localisation with and the activation of LRP5 and LRP6. The Wnt/FZD/LRP signalling drives the dishevelled-mediated attenuation of the β-catenin inhibitory complex (CK-1/Axin/APC/GSK-3), facilitating the accumulation of β-catenin and its translocation to the nucleus, wherein it associates with co-transcription factors, TLF and LEF, to induce pro-fibrotic gene expression.
2. The Non-Canonical Pathway: HA/CD44/EGFR
A principal mediator of a non-canonical differentiation pathway in human myofibroblasts is HA, a glycosaminoglycan (GAG) formed of repeating d-glucuronic acid and N-acetyl-glucosamine disaccharide units. HA is synthesised by three HA-synthase (HAS) isoenzymes (HAS1, HAS2, HAS3) [10][55], and is metabolised by hyaluronidase (HYAL) enzymes (HYAL1, HYAL2, HYAL4) [11][56]. HA accumulation occurs simultaneously with upregulated HYAL1 and HYAL2 expression, but both these HA degrading enzymes have diminished expression in myofibroblasts. A subsequent decrease in HA degradation may also favour the accumulation of HA in fibrotic disease [12][57]. HAS2-synthesised HA is widely considered to be an important mediator of fibroblast–myofibroblast differentiation and EMT processes [13][14][15][16][17][58,59,60,61,62]. HAS2-synthesised linear HA forms a thickened pericellular coat that surrounds the activated fibroblast. This crosslinked and thickened HA pericellular coat, or glycocalyx, has been shown to be an essential structural element for human fibroblast–myofibroblast differentiation and phenotypic maintenance [14][18][59,63]. The HA pericellular coat remains thin in fibroblast phenotypes resistant to TGF-β1-driven differentiation, such as oral mucosal fibroblasts [19][64] and aged/senescent fibroblasts [13][58]. Interestingly, an enriched pericellular HA microenvironment is synthesised by foetal fibroblasts during foetal wound healing.
The HA pericellular coat is anchored to the fibroblast cell surface by the CD44 receptor [20][21][65,66], and crosslinked by hyaladherins [14][59]. Association with CD44 and other hyaladherins orchestrates the HA pericellular coat and results in the activation of downstream extracellular–intracellular signal cascades, shown to be prominent in disease pathogenesis including fibrosis, cancers, and inflammatory diseases [22][23][67,68]. CD44 exists as multiple alternatively spliced variants and all CD44 variants (CD44v), as well as the standard form (CD44s), possess HA binding motifs within their extracellular head domain. CD44v have variability within alternatively spliced exons of the extracellular stem region [24][25][69,70]. Studies have shown that silencing CD44 using small interfering RNA (siRNA) prevented the downstream intracellular signalling required for human fibroblast–myofibroblast differentiation and inhibited HA pericellular coat formation [26][27][71,72]. These studies concluded that HA/CD44 association was essential for human myofibroblast differentiation, with recent research showing that CD44s expressed on the surface of human fibroblasts was the dominant player in regulating HA coat-mediated myofibroblast differentiation [28][73]. HA-crosslinking hyaladherins include inter-α-inhibitor (IαI) heavy chains (HC), which are localised by association with the tumour necrosis factor-stimulated gene (TSG)-6 [29][30][74,75]. The increased presence of HA-associated HC and TSG-6 complexes have been associated with the pathology of multiple inflammatory diseases including arthritis, asthma, and some cancers [31][76]. Martin et al. identified that IαI-HC5 was covalently bound to chondroitin sulphate on bikunin and was essential for the formation of HA pericellular coats in myofibroblasts. Increased expression of TSG-6 following TGF-β1 activation of fibroblasts facilitated the transfer of HC5 to HA by TSG-6 catalysis, in a metal ion-dependent manner [32][77].
HA binding to CD44 plays a pivotal role in the intracellular signalling required for human fibroblast–myofibroblast differentiation [13][18][27][58,63,72]. CD44 was shown to be present throughout the membranes of fibroblasts but clustered into localised populations within myofibroblast membranes. These distinct areas were identified to be cholesterol-rich lipid rafts, and ‘locked’ clusters of CD44 within lipid rafts were shown to be dependent on HA binding and crosslinking into pericellular coats [27][72]. HA coat removal released CD44 from lipid rafts and re-enabled their movement through the membrane, but also inhibited downstream signalling cascades [18][63]. Further investigation revealed that CD44 was co-localised with epidermal growth factor (EGFR) within membrane lipid raft domains, which resulted in EGFR activation, and the phosphorylation of downstream effectors and transcription factors, ERK1/2 and calcium calmodulin kinase (CaMK)-II (CaMKII). The chemical inhibition of ERK1/2 or CaMKII prevented myofibroblast differentiation, and only when TGF-βRII and CD44/EGFR signalling were active could myofibroblast differentiation occur [27][72]. Both ERK1/2 and CaMKII were phosphorylated in a biphasic manner, with early and late-phase activation. Early activation of ERK1/2 (<5 min) was thought to be associated with eventual fibroblast–myofibroblast differentiation, whereas later activation (>30 min) was suggested to play a role mediating cellular proliferation responses [27][72] (Figure 12B). However, signalling functions appear to be cell-dependent as early, but not late, ERK1/2 signalling peaks were observed in TGF-β1-stimulated non-scarring oral mucosal fibroblasts; these cells exhibited an anti-proliferative response in a HA/CD44-independent manner [20][65].
3. The Wnt/β-Catenin Pathway
The Wnt signalling pathway is well described in its vital roles during embryogenesis and determination of cell fate, differentiation, proliferation, and apoptosis. Carefully orchestrated Wnt signalling has been found to be essential for tissue homeostasis, whilst the dysregulation of the Wnt pathway can result in pathogenesis [33][34][79,80]. Wnt signals simultaneously through the co-association of Frazzled receptors and low-density lipid protein co-receptors (LRP). The fate of β-catenin, an important regulator of Wnt signalling, is regulated by Dickkopf-related protein-1 (DKK1). DKK1 inhibits Wnt signalling by activating downstream signalling complexes, and results in β-catenin degradation. When DKK1 is not present, Wnt signalling leads to β-catenin translocation to the nucleus, its association with co-transcription factors, and the subsequent activation of Wnt-associated pro-fibrotic genes [33][35][36][78,79,81] (Figure 12C). The activation of Wnt signalling was implicated in the fibrogenesis of multiple organs [37][82]. Wnt-1 and Wnt-10b were noted to be overexpressed, whereas DKK1 was decreased, which led to the increased nuclear accumulation of β-catenin observed in human tissue samples from systemic scleroderma (SSc), idiopathic pulmonary fibrosis (IPF), and liver cirrhosis [37][82]. Wnt signalling stimulated by TGF-β1-induced inhibition of DKK1 was shown to induce myofibroblast differentiation, up-regulate the release of ECM components (notably collagens) and induce fibrosis [37][38][39][82,83,84]. In addition, crosstalk between the TGF-β/Smad pathway and the Wnt/β-catenin pathway is suggested to be a prominent mechanism in the development of hypertrophic and keloid scars [40][85].
 Discoidin domain receptor (DDR)1 is a mediator of stromal–epithelial interactions, and belongs to the family of collagen receptors [73][116] overexpressed in pro-fibrotic keloid fibroblasts [74][117]. DDR1 mediates collagen contraction and stiffness, thereby indirectly mediating pro-fibrogenic responses that are largely independent from collagen binding to integrins [75][118]. Additionally, the spatial and structural properties of the local 3D collagen microarchitecture can distinctly affect fibroblast and myofibroblast activity. Collagen fibril alignment and diameter were shown to affect fibroblast contractility and migration via alteration in integrin clustering and the stability of adhesion sites [76][119]. Seo and colleagues suggested that collagen fibre thickness held more pertinence over dictating myofibroblast differentiation independently from collagen quantity [77][120]. Furthermore, collagen-rich ECM stiffening can also be potentiated by the myofibroblast and inflammatory cell production of collagen crosslinking enzymes, such as lysyl oxidases [47][78][11,90]. CD147, also known as extracellular matrix metalloproteinase inducer (EMMPRIN), is a well-established matrix metalloproteinase (MMP)-inducer that is rapidly becoming understood to mediate multiple cellular responses. The linked association of CD147 with integrin α6β1 was observed during increased metastasis in human hepatoma cells [79][121], suggesting a role for CD147 in modulating integrin α6β1 associations, with matricellular protein CCN1, and mechano-sensitivity to senescence or apoptosis [80][122]. Interestingly, the nullification of the extracellular domain of CD147 reversed associations with integrin β subunits and FA sites, suggesting that the extracellular domain of CD147 binds to integrins to regulate the mechanical tension of the ECM [79][81][121,123]. The roles of these mechanotransducer proteins in stress relay, which is involved in myofibroblast differentiation, will become clearer with continued investigation.
Increasing evidence suggests that myofibroblasts have the capacity for classical CaMK-myosin light chain kinase (MLCK)-dependent smooth muscle cell contraction mechanisms [82][124] and contractile activation by the RhoA/ROCK/myosin light chain phosphatase (MLCP) pathway [44][83][84][28,125,126], highlighting a key difference between the smooth muscle cell and myofibroblast contractile mechanisms. It was recently discovered that HYAL2, previously thought to only possess HA catalytic activity, was shown to have non-enzymatic functions [24][85][86][69,127,128]. Following TGF-β1 activation, enhanced HAS2 expression by fibroblasts leads to increased HA accumulation within the ECM observed in 3D matrices in vitro [87][129] and in fibrosis progression in vivo [88][130]. However, HYAL2 was shown to re-localize to the cytoplasm and align along the actin cytoskeleton in myofibroblasts [86][128], which may contribute to increasing extracellular HA accumulation. The cytoskeleton-aligned HYAL2 was associated with α-SMA, RhoA, and MLCK. The silencing of HYAL2 did not prevent but only delayed myofibroblast differentiation. The presence of HYAL2 was shown to accelerate RhoA/MLCK phosphorylation and cellular contractility, suggesting a role in the mechanotransduction and the orchestration of key cytoskeletal and FA-related proteins [86][128]. These early findings suggest that there may be complex orchestration roles that HYAL2 plays in cytoskeletal reassembly.
Other mediators of mechanotransduction and promising targets in mediating fibroblast–myofibroblast differentiation include myocardin-related transcription factor (MRTF) [89][90][131,132], Yes-associated protein (YAP) [91][92][93][94][95][133,134,135,136,137], cadherins [96][138], and Notch [97][139]. In the case of these multifunctional proteins, further exploratory research will help establish the feasibility of targeting their activity to interfere with myofibroblast differentiation or function.
Discoidin domain receptor (DDR)1 is a mediator of stromal–epithelial interactions, and belongs to the family of collagen receptors [73][116] overexpressed in pro-fibrotic keloid fibroblasts [74][117]. DDR1 mediates collagen contraction and stiffness, thereby indirectly mediating pro-fibrogenic responses that are largely independent from collagen binding to integrins [75][118]. Additionally, the spatial and structural properties of the local 3D collagen microarchitecture can distinctly affect fibroblast and myofibroblast activity. Collagen fibril alignment and diameter were shown to affect fibroblast contractility and migration via alteration in integrin clustering and the stability of adhesion sites [76][119]. Seo and colleagues suggested that collagen fibre thickness held more pertinence over dictating myofibroblast differentiation independently from collagen quantity [77][120]. Furthermore, collagen-rich ECM stiffening can also be potentiated by the myofibroblast and inflammatory cell production of collagen crosslinking enzymes, such as lysyl oxidases [47][78][11,90]. CD147, also known as extracellular matrix metalloproteinase inducer (EMMPRIN), is a well-established matrix metalloproteinase (MMP)-inducer that is rapidly becoming understood to mediate multiple cellular responses. The linked association of CD147 with integrin α6β1 was observed during increased metastasis in human hepatoma cells [79][121], suggesting a role for CD147 in modulating integrin α6β1 associations, with matricellular protein CCN1, and mechano-sensitivity to senescence or apoptosis [80][122]. Interestingly, the nullification of the extracellular domain of CD147 reversed associations with integrin β subunits and FA sites, suggesting that the extracellular domain of CD147 binds to integrins to regulate the mechanical tension of the ECM [79][81][121,123]. The roles of these mechanotransducer proteins in stress relay, which is involved in myofibroblast differentiation, will become clearer with continued investigation.
Increasing evidence suggests that myofibroblasts have the capacity for classical CaMK-myosin light chain kinase (MLCK)-dependent smooth muscle cell contraction mechanisms [82][124] and contractile activation by the RhoA/ROCK/myosin light chain phosphatase (MLCP) pathway [44][83][84][28,125,126], highlighting a key difference between the smooth muscle cell and myofibroblast contractile mechanisms. It was recently discovered that HYAL2, previously thought to only possess HA catalytic activity, was shown to have non-enzymatic functions [24][85][86][69,127,128]. Following TGF-β1 activation, enhanced HAS2 expression by fibroblasts leads to increased HA accumulation within the ECM observed in 3D matrices in vitro [87][129] and in fibrosis progression in vivo [88][130]. However, HYAL2 was shown to re-localize to the cytoplasm and align along the actin cytoskeleton in myofibroblasts [86][128], which may contribute to increasing extracellular HA accumulation. The cytoskeleton-aligned HYAL2 was associated with α-SMA, RhoA, and MLCK. The silencing of HYAL2 did not prevent but only delayed myofibroblast differentiation. The presence of HYAL2 was shown to accelerate RhoA/MLCK phosphorylation and cellular contractility, suggesting a role in the mechanotransduction and the orchestration of key cytoskeletal and FA-related proteins [86][128]. These early findings suggest that there may be complex orchestration roles that HYAL2 plays in cytoskeletal reassembly.
Other mediators of mechanotransduction and promising targets in mediating fibroblast–myofibroblast differentiation include myocardin-related transcription factor (MRTF) [89][90][131,132], Yes-associated protein (YAP) [91][92][93][94][95][133,134,135,136,137], cadherins [96][138], and Notch [97][139]. In the case of these multifunctional proteins, further exploratory research will help establish the feasibility of targeting their activity to interfere with myofibroblast differentiation or function.
4. Mechanotransduction
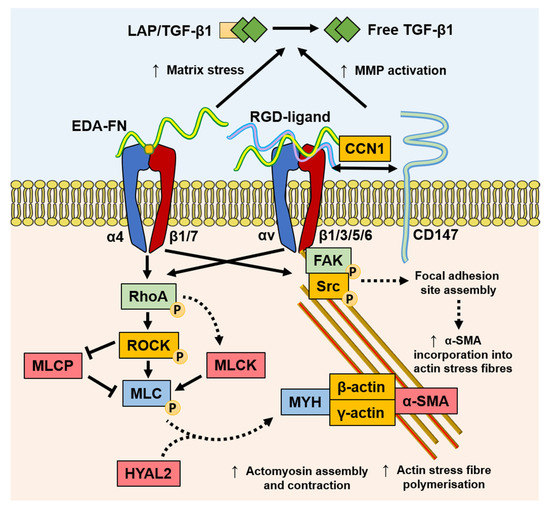
Mechanotransduction is the ability of stress force to convert extracellular to intracellular signalling. Fibroblasts can perceive external forces (mechanoperception) through their fibronexus structures in vivo or mature focal adhesion (FA) structures in vitro [41][42][86,87]. ECM rigidity determines the size of the cell’s FAs, or ‘anchors’, which in turn limit the level of tension generated within intracellular stress fibres. Only when substrate stiffness permits the formation of mature FAs (8–30 μm), and the generation of approximately four-fold greater stress compared with usual FAs (2–6 μm), does α-SMA become incorporated into pre-existing cytoplasmic β-actin stress fibres [43][88]. Thus, the myofibroblast phenotype is mechanosensitive. The transition of fibroblasts to the proto-myofibroblast state was suggested to be related to increased microenvironment stiffness [43][44][28,88]. TGF-β1 is locked within the ECM by latency-associated peptide (LAP) and latent TGF-β1-binding protein (LTBP), and is released by proteolysis or integrin-dependent mechanotransduction [45][89]; resulting in a feedback loop of pro-fibrotic fibroblast activity and increased ECM stiffness [46][47][48][49][29,90,91,92]. In a recent study, the balance of ECM composition, elasticity, and TGF-β1 signalling was shown to govern fibroblast phenotypic heterogeneity and give rise to distinct, but overlapping, fibroblast subsets [50][93]. Indeed, Kollmannsberger and colleagues showed that within 3D microtissues grown in vitro, fibroblasts transitioned to proliferative myofibroblasts at the growth front, where a high degree of tensile force and stretched FN fibres were present. As the tissue matrix matured into collagen-rich ECM with low FN fibre tension, more fibroblasts were present, suggesting that the myofibroblast phenotype stabilised by tensile forces could revert to a fibroblast phenotype by low tensile force [51][94]. These studies suggest that substrate stiffness is a key determinant of fibroblast and myofibroblast plasticity, and that reducing excessive mechanotransduction may be an option in controlling myofibroblast presence. Myofibroblasts generate force through stress fibre contraction, and this is transmitted to the ECM via FAs containing transmembrane receptors, typically integrins [42][52][87,95], as summarised in Figure 23. Integrins mediate cell–cell, and especially, cell–ECM interactions, and are prominently involved in the initiation, maintenance, and resolution of fibrosis. EDA–FN is synthesised by fibroblasts and various other cell types [53][54][96,97]. The EDA–FN splice variant can only be detected during tissue repair [55][56][98,99], fibrosis [57][100], tumour development [58][59][60][101,102,103], and transiently during embryogenesis [61][104]. The accumulation of EDA–FN has been observed in multiple fibrotic disorders, including lung, liver, and skin [57][62][63][64][100,105,106,107]. EDA–FN promotes myofibroblast differentiation by orchestrating LAP/LTBP release and activation of TGF-β1 [65][108], increasing matrix stress–strain tension [66][67][109,110], and by activating mechanotransducer-integrin signalling via FAK activation [68][69][70][71][111,112,113,114]. Research by Shinde et al. [72][115] and Kohan et al. [69][112] demonstrated that fibroblast-expressed integrins, α4β1 (VLA-4) and α4β7 (LPAM-1), mediate different roles in myofibroblast formation, respectively. Integrin α4β1/EDA–FN promoted ECM synthesis and stiffening [72][115], whereas integrin α4β7/EDA–FN induced myofibroblast stress fibre formation and contractility [69][112]. The combined exclusivity of EDA–FN expression and the prominent roles it plays in driving the myofibroblast phenotype have highlighted the protein as an attractive target for anti-fibrotic interventional therapies.Figure 23. Mechanisms of mechanotransduction implicated in myofibroblast differentiation and function. TGF-β1 stimulates an increased expression of EDA–FN. The EDA segment contains the EDGIHEL motif, which binds to the integrin α4β1/α4β7 receptor cleft, initiating signalling through RhoA/MLCK and FAK. RhoA signalling results in myosin light chain (MLC) promotion of myosin (MYH)/actin assembly and stress fibre polymerisation, which is augmented by HYAL2 that relocates from the membrane to the cytoplasm. RGD motif containing ligands, such as FN and collagens, bind integrins and facilitate the formation of focal adhesion sites, increasing contractile force and α-SMA incorporation into actin stress fibres. In addition, the increased matrix stress generated by EDA–FN and increased MMP activation from CD147 recruitment exacerbates active TGF-β1 release from the extracellular LAP complex.