Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Antonio Bianchi and Version 2 by Alfred Zheng.
Hypophysitis, a rare inflammatory disorder of the pituitary gland, has seen an uptick in reported cases in recent years. Immune checkpoint inhibitors induce hypophysitis (IIHs): IIHs is an increasingly frequent toxicity of in patients on treatment with inhibitors targeting cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death-1 (PD-1).
- hypophysitis
- autoimmune
1. Introduction
Hypophysitis, a rare inflammatory disorder of the pituitary gland, has seen an uptick in reported cases in recent years. This surge can be attributed to intensified research into the etiological causes of hypopituitarism, enhancements in radiological techniques, and the clinical use of drugs that may favor the occurrence of immune-related adverse events [1][2][3][4][5][1,2,3,4,5].
Hypophysitis exhibits significant heterogeneity and variability in its etiology. The most prevalent form is primary autoimmune hypophysitis (PAH), which is often idiopathic [4][5][4,5]. Moreover, hypophysitis has been identified as a clinically significant endocrine toxicity in patients undergoing treatment with immune-checkpoint inhibitors (ICIs). ICIs, by bolstering immune tumor surveillance, are approved for the treatment of various malignancies [1][2][1,2]. Furthermore, hypophysitis also appears to be prompted by a paraneoplastic autoimmune reaction. The presence of diverse ectopic proteins expressed in tumors can induce the formation of autoantibodies and autoreactive cytotoxic T cells [6][7][8][6,7,8]. Lastly, the recent literature has reported cases of hypophysitis linked to the SARS-CoV-2 virus, as well as to COVID-19 vaccination [5][9][10][11][12][5,9,10,11,12].
While the clinical aspects, diagnosis, and management of hypophysitis have been extensively studied, the etiopathogenesis and molecular mechanisms underlying hypophysitis remain largely elusive [4][5][13][14][15][4,5,13,14,15]. Acquiring a deeper understanding of the etiopathogenesis and discovering biomarkers for hypophysitis could potentially improve clinical practice by guiding more efficacious therapeutic choices [15].
2. Immunotherapy Induced Hypophysitis
2.1. Pathogenesis of Immunotherapy Induced Hypophysitis
Hypophysitis is an increasingly frequent endocrine toxicity of clinical significance observed in patients on treatment with ICIs, particularly monoclonal antibodies (mAbs) that target cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death-1 (PD-1) [1][2][1,2]. Nonetheless, there are limited data available on the molecular mechanism underlying this disorder.
The varying incidence rates of ICI-induced hypophysitis (IIH) have been attributed to the different mechanisms of action of the ICI: antibodies that block CTLA-4 enhance T-cell priming, while those that block PD-1/PD-L1 seem to enhance an existing CD8 T-cell response [2][16][2,59].
CTLA-4 is expressed on the cell surface of active CD4-positive and CD8-positive T cells. The primary function of the CTLA-4 pathway is to drain lymph nodes where naïve T-cells are primed by exposure to tumor antigens (presented by antigen-presenting cells) and become activated [17][60]. CTLA-4 binds CD80 and CD86, which are expressed on the cell surface of APCs, with higher affinity and avidity than CD28 [18][61]. The engagement of CTLA-4 with CD80 and CD86 mitigates the immune response [19][62]. ICIs inhibit the CTLA-4 pathway, leading to overactivation of T-lymphocytes. This overactivation may predispose to the onset of autoimmune disease, such as IIH. The increased incidence of hypophysitis in patients treated with anti-CTLA-4 has also been linked to the potential ectopic expression of CTLA-4 at the pituitary gland level. This ectopic expression may act as an autoantigen, which could trigger an autoimmune reaction by anti-CTLA-4 antibodies in cancer patients undergoing ICI treatment [16][59] (Figure 1).
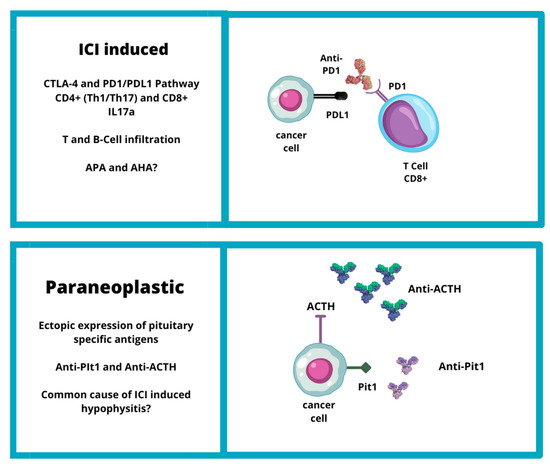
Figure 1. Etiopathogenesis of IIH (CTLA4: cytotoxic T-lymphocyte-associated protein 4; PD1: programmed cell death protein-1; PDL1: programmed death-ligand 1; ACTH: adrenocorticotropic hormone; Pit1: pituitary transcription factor 1).
Regarding PD-1 targeted ICIs, PD-1 is an inhibitory receptor expressed mainly on activated CD8-positive T-lymphocytes [2][20][2,63]. PD-1 is triggered by PD-ligands 1 and 2 (PD-L1 and PD-L2, respectively), which are constitutively expressed on tumor cells [21][22][64,65]. The binding of PD-1/PD-L1 suppresses the activity of T-cells [23][66] promotes the conversion of T-helpers into T-regulatory cells [24][67] and activates pro-survival signaling pathways in cancer cells, leading to resistance to cytotoxic T-lymphocytes [25][68]. According on the inhibition of this mechanism, treatment with ICI targeting PD-1 and PD-L1 is associated with a high frequency of autoimmune disorders, including endocrine toxicity [1].
Recent findings suggest that cytokines, in addition to T-cell-mediated cytotoxicity, play a crucial role in IIHs. In mouse models of autoimmune hypophysitis, infiltrates of CD4 T cells with T helper cell 1 and 17 (Th1/Th17) and T helper cell 17 (Th17) cytokine profiles were observed in the pituitary gland. Furthermore, transcriptome analysis of IIHS demonstrated the predominant expression of interleukin 17A (IL-17A), CD4, and MHC class II antigens [26][69]. Chalan et al. isolated RNA from the formalin-fixed paraffin-embedded pituitary specimens of 16 hypophysitis patients (three of whom had IIHs). All three secondary hypophysitis patients showed detectable IL-17A levels and other cytokines were not detected [27][24].
B-cell infiltration has been observed in IIHs, suggesting that antibody-mediated pituitary injury may contribute to the pathogenesis of IIHs as in autoimmune hypophysitis [26][28][51,69]. The prevalence of APAs and AHAs during anti-PD-1/anti-PD-L1 therapy is higher than in healthy individuals; in some patients, APA/AHA positivity has been observed after just 9 weeks of immunotherapy [29][70]. APAs, measured by indirect immunofluorescence, are a surrogate marker of the presence of autoimmunity against the pituitary gland and are detected more frequently in some pituitary diseases, particularly biopsy-proven hypophysitis. Although APAs are negative at baseline, one study demonstrated that APAs became positive at the onset of ipilimumab (anti-CTLA-4 antibody)-induced hypophysitis in all examined patients [30][71].
Lastly, the prevalence of hypophysitis in patients treated with anti-PD-1 and anti-PD-L1 increases when these ICIs are associated with anti-CTLA-4 antibodies or when patients have pre-existing autoimmune or inflammatory disorders. One proposed mechanism is an increase in T-cell activity against shared tumor and normal tissue antigens due to pre-existing antibodies or inflammatory cytokines. However, studies on the detection of antibodies in cancer patients treated with ICIs, particularly PD-1 and PD-L1 inhibitors, are rare and have primarily focused on the detection of APA, not AHA. Recently, APA was detected in two out of four cancer patients with hypophysitis related to anti-PD-L1 or anti-PD-1 treatment [16][28][51,59].
2.2. Genetic Factors of ICI-Induced Hypophysitis
The pathogenesis of IIHs involves various mechanisms, among which genetic factors play a crucial role. It is well known that genetic polymorphisms of CTLA-4 and PD-1 genes can increase the risk of developing autoimmune diseases, including IIHs [31][32][72,73]. These polymorphisms may not alter the CTLA-4 amino-acid sequence but can affect the affinity for CTLA-4 mAbs, thereby increasing the risk of immunotherapy-induced autoimmune disorders [31][72].
HLA alleles may also be involved in the onset of IIHs, with several HLA haplotypes being associated with autoimmune diseases [8]. In particular, a Japanese study showed that HLACw12 and HLA-DR15 were significantly associated with anti-CTLA-4 related hypophysitis, whereas HLA-DQB106:01, HLA-DPB109:01, and HLA-DRB5*01:02 were significantly associated with anti-PD-1-related hypophysitis [26][69]. Another study found that HLA-Cw12, HLA-DR15, HLA-DQ7, and HLA-DPw9 were significantly more prevalent in patients with immunotherapy induced central hypoadrenalism [8][30][8,71]. This HLA association presents a possible alternative mechanistic hypothesis in contrast to the proposed hypothesis that ICIs hypophysitis is due to direct binding of CTLA-4 inhibitors to pituitary cells [33][74].
Additionally, copy number variations (CNVs) and small variations (VARs) may also be associated with the occurrence of IIHs. A study analyzing 95 melanoma patients treated with ICIs found that genes affected by VARs associated with hypophysitis include SMAD3, PRDM1, and IL1RN, while genes affected by CNVs related to the occurrence of hypophysitis are TERT, SMAD3, JAK2, PRDM1, FAN1, CD274, and UNG [29][70].
Lastly, congenital forms of isolated ACTH deficiencies commonly involve the T-PIT gene, also known as the TBX19 gene, and the POMC gene, which encodes the melanocortin protein [30][71].
2.3. Paraneoplastic Syndrome Hypothesis
Recently, a hypothesis has emerged that IIHs may be linked to a paraneoplastic syndrome triggered by ectopic expression of pituitary specific antigens. Two new clinical entities have been identified: anti-PIT-1 hypophysitis and paraneoplastic autoimmune isolated ACTH deficiency [6] (Figure 1).
Bando et al. described the mechanism of a new kind of hypophisitis: anti-PIT-1 hypophysitis. It was described as paraneoplastic autoimmune hypophysitis due to ectopic expression of PIT-1 or pituitary hormones in complicated tumors that evokes autoimmunity against pituitary cells, leading to the production of anti-Pit-1 autoantibodies and autoreactive cytotoxic T cells. The presence of autoimmunity directed against PIT-1 could explain the pituitary hormone abnormalities in some cancer patients and that tumor tissues sometimes exhibited ectopic expression of pituitary antigens and hormones, such as POMC [6]. In addition, in an autopsy specimen, it was observed that the PIT-1 epitope presented with MHC class I antigen on the surface of patient pituitary cells [34][75]. Furthermore, the presence of a common underlying mechanism between “paraneoplastic autoimmune hypophysitis” and PD-1/PDL-1 inhibitor-related hypophysitis was suggested. Actually, some cases of PD-1/PD-L1 inhibitor-related hypophysitis could be caused by these paraneoplastic autoimmune mechanisms [6].
Similarly, in paraneoplastic autoimmune isolated ACTH deficiency, ectopic expression of cancer cells in ACTH can promote the immune response, leading to the synthesis of anti-ACTH antibodies that can also act on pituitary corticotroph cells and cause central hypoadrenalism [28][51]. Recent research suggests that IIHs and ACTH deficits may be considered two different endocrine toxicities with manifestation of the same clinical entity. A study conducted on 62 cancer patients treated with ICIs found that the prevalence of APAs was similar among the five patients who developed an IIH (APA positivity: 80%) and in those who developed an ACTH deficit (APA positivity: 88.2%) entity [8]. Kobayashi et al. (2021) reported that APAs recognized ACTH-secreting cells in all patients with positive APAs, not only at baseline but also at the onset of ICI-induced isolated ACTH deficiency. Moreover, in some patients, APAs recognized pituitary cells secreting other hormones such as TSH, FSH, LH, GH, and/or PRL at the onset of ICI-induced isolated ACTH deficiency [28][30][51,71].