Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Luisa Maia and Version 5 by Rita Xu.
Nitric oxide radical (NO) is a signalling molecule involved in several physiological and pathological processes and a new nitrate-nitrite-NO pathway has emerged as a physiological salvage pathway that operates under challenging conditions, when the "classic" L-arginine-dependent NO synthases are hindered. To catalyse the reduction of nitrite to NO, mammalian cells can use different metalloproteins that are present in cells to perform other functions (moonlighting). Among the so far identified ''non-dedicated nitrite reductases'', the molybdenum-containing enzymes stand out as very efficient NO synthases due to their well know ability to catalyse oxygen atom transfer reactions.
- xanthine oxidase
- aldehyde oxidase
- molybdenum enzyme
- nitrite
- cell signalling
- moonlighting
1. Context—I: The Molybdenum Side
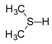
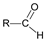
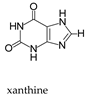
Molybdenum is essential to the great majority of organisms [1][2][3][4][1,2,3,4], from archaea and bacteria to higher plants and mammals, being present in the active site of enzymes that catalyse redox reactions targeting nitrogen, sulfur and carbon atoms of key metabolites [5][6][7][8][5,6,7,8]. Molybdenum-containing enzymes are responsible for the biological handling of dinitrogen (Nitrogenase, Equation (1)), nitrate (Nitrate reductase (NaR), Equation (2)), sulfite (Sulfite oxidase (SO), Equation (3)), dimethylsulfoxide (dimethylsulfoxide reductase (DMSOR), Equation (4)), aldehydes (Aldehyde oxidase (AO), Equation (5)), xanthine (Xanthine oxidase (XO), Equation (6)) and carbon dioxide (Carbon dioxide dehydrogenase, Equation (7), and Formate dehydrogenase, Equation (8)), just to mention a few examples from the more than 50 enzymes presently known.
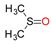
 + 2e
+ 2e
 + H2O →
+ H2O → 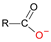 (H+) + 2e− + 2H+
(H+) + 2e− + 2H+
 + H2O →
+ H2O → 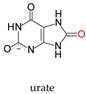 (H+) + 2e− + 2H+
(H+) + 2e− + 2H+
N≡N + 8H+ + 8e− + 16MgATP → 2NH3 + H2 + 16MgADP + 16Pi
ONO2− + 2e− + 2H+ → NO2− + H2O
SO22− + H2O → OSO22− + 2e− + 2H+
+ 2e+ 2e
−
+ 2H
+ → 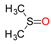 + H2
+ H2
+ H2 →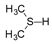
+ H2
O
+ H2O → (H+) + 2e− + 2H+ + H2O → (H+) + 2e− + 2H+ CO + H2O → OCO + 2e− + 2H+
HCOO− → CO2 + 2e− + H+
With the single (as far as is presently known) exception of nitrogenase [9][10][11][12][9,10,11,12], molybdenum is found coordinated by the cis-dithiolene group (–S–C=C–S–) of one or two molecules of a pyranopterin cofactor (Figure 1). In a parallel situation to the haem ring, this unique cofactor is not an “innocent scaffold” and it is considered to be co-responsible to modulate the active site reactivity, besides acting as a “wire” to conduct the electrons to, or from, the other redox-active centres of the enzyme (intramolecular electron transfer, when this is the case) [13][14][15][16][17][18][19][20][13,14,15,16,17,18,19,20]. In addition to the pyranopterin cofactor, the molybdenum ion is coordinated by oxygen and/or sulfur and/or selenium terminal atoms and/or by enzyme-derived amino acid residues (Figure 1), which are also expected to have key roles in catalysis (although their individual roles are not yet understood in many molybdoenzymes). To introduce order in such a diversity of molybdenum compositions and (try to) rationalise the catalytic features, the molybdoenzymes were classified into three large families, denominated after one benchmark enzyme, as indicated in Figure 1 [5]: XO family, SO family and DMSOR family.
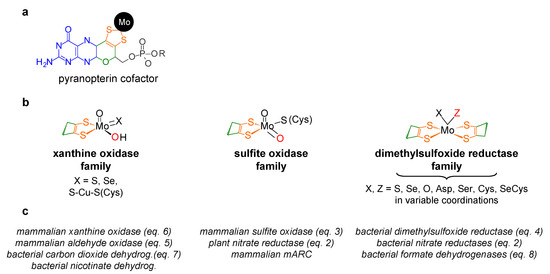
Figure 1. Structures of the molybdenum centres of the three families of molybdoenzymes. (a) Pyranopterin cofactor structure. The molecule is formed by pyrano (green)-pterin (blue)-dithiolene (orange) moieties. In eukaryotes, the cofactor is found in the simplest monophosphate form (R is a hydrogen atom; in gray), while in prokaryotes, it is most often found esterified with several nucleotides (R can be one cytidine monophosphate, guanosine monophosphate or adenosine monophosphate). (b) Structures of the molybdenum centres, in the oxidised form, of the three families of molybdoenzymes. For simplicity, only the cis-dithiolene group of the pyranopterin cofactor is represented. The labile oxygen position is indicated in red. It should be noted that the XO family member carbon monoxide dehydrogenase (X = S-Cu-S(Cys)) is suggested to harbour a labile Mo=O group and not Mo-OH. (c) Selected examples of enzymes from each family (mARC, mitochondrial amidoxime reducing component).
These molybdenum centres have been exploited by living organisms to carry out different reaction types [5][6][7][8][5,6,7,8], many of which are oxygen atom transfer reactions (OAT), where an oxygen atom is transferred from water to product -oxygen atom insertion (OAT-I) (Figure 2, violet arrows)- or from substrate to water -oxygen atom abstraction (OAT-A) (Figure 2, red arrows).
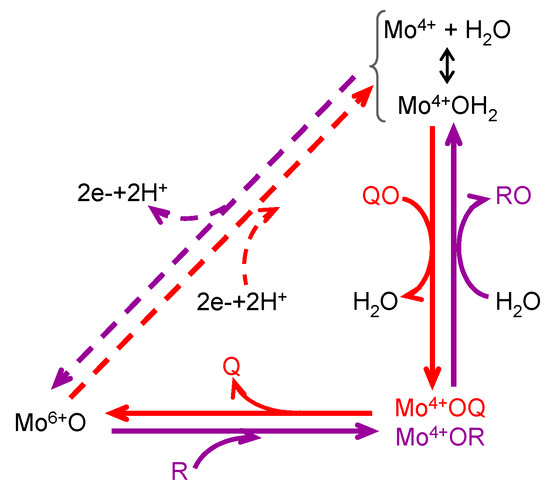
Figure 2. General mechanism of molybdoenzymes-catalysed OAT. The molybdenum role as electron acceptor/donor and as direct oxygen donor to substrate/oxygen acceptor from product is highlighted. OAT-I is represented in violet; e.g., for SO (Equation (3)), R would be sulfite and RO is sulfate; reduction of the enzyme physiological partner/regeneration of the enzyme active site is represented in dashes. OAT-A is represented in red; e.g., for NaR (Equation (2)), QO would be nitrate and Q is nitrite; oxidation of the enzyme physiological partner/regeneration of the enzyme active site is represented in dashes. It should be noted that catalysis can take place without the occurrence of the dashed reduction/oxidation.
The reaction mechanism of molybdoenzymes-catalysed OAT-I and OAT-A is presently well established and can be illustrated with the eukaryotic SO (Equation (3)) and NaR (Equation (2)) catalytic cycles (two enzymes from the SO family). Briefly, in SO [21][22][23][24][25][26][27][28][29][30][31][32][33][34][21,22,23,24,25,26,27,28,29,30,31,32,33,34] (Figure 3a), catalysis is initiated at the oxidised molybdenum centre, where its equatorial labile oxido group (Mo6+=Oequatorial) is (nucleophilically) attacked by the sulfite lone-pair of electrons, resulting in the formation of a reduced, covalent Mo4+-O-SO3 intermediate. After cleavage of the Mo-O(substrate)equatorial bond, sulfate is released (oxidation half-reaction). The Mo4+-OH(2) centre thus formed is then re-oxidised by two electrons (with the eventual reduction of the SO physiological partner—reduction half-reaction) to regenerate the initial Mo6+=O centre. The mechanism of OAT-A of NaR is suggested to be the reverse of the SO one (Figure 3b) [35][36][37][38][39][40][41][35,36,37,38,39,40,41], starting with the reduction of the molybdenum centre (two electrons provided by the NaR physiological partner—oxidation half-reaction). Nitrate binding yields the covalent intermediate Mo4+-O-NO2, which, after cleavage of the O-N bond and release of nitrite (reduction half-reaction), regenerates the Mo6+=O core. Hence, in a simplistic way, the molybdenum role in OAT is to accept/donate the necessary electrons for these redox reactions and to act as the direct oxygen donor to substrate/oxygen acceptor from product (Figure 2).
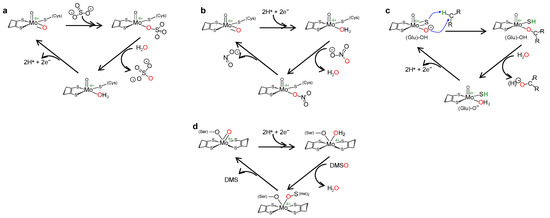
Figure 3. Simplified mechanism of OAT catalysed by eukaryotic SO (a), NaR (b), XO (c) and prokaryotic DMSOR (d). See text for details.
This general OAT mechanism (Figure 2) is suggested to be followed by other molybdoenzymes, from other families, but with some variations imposed by the respective substrate nature. This is the case of the XO-catalysed xanthine hydroxylation to urate (Equation (6); OAT-I) a reaction more “complex” than the “simple” OAT of SO and NaR, because it requires the cleavage of the stable C(8)-H bond of xanthine for the oxygen to be inserted. To catalyse the xanthine hydroxylation, the molybdenum active site of XO holds an equatorial labile oxido group that is the direct source of the oxygen atom to be inserted into the xanthine C(8) (in parallel to SO). However, in the place of the S-coordinated cysteine residue of SO, XO active site harbours a terminal sulfido group to accept the xanthine H(8) and make its labile oxide group capable of carrying out a nucleophilic attack (instead of undergo a nucleophilic attack as in SO). Briefly, XO catalysis (Figure 3c) [42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][42,43,44,45,46,47,48,49,50,51,52,53,54,55,56] is initiated with the activation of the equatorial labile oxido group by a conserved glutamate residue, to form the catalytically competent Mo6+(=S)-O− centre. This makes a nucleophilic attack into xanthine C(8), while the sulfido group accepts the xanthine H(8) as a hydride, leading to the formation of a reduced, covalent intermediate Mo4+-O-C-R(-SH) (where R represents the remainder of the xanthine molecule). After hydrolysis of the Mo-O(xanthine)equatorial bond, urate is released (oxidation half-reaction) and the Mo4+(SH)-OH(2) centre formed is re-oxidised by two electrons (with the eventual reduction of the XO physiological partner—reduction half-reaction) to regenerate the initial Mo6+(=S)-O− centre.
The enzymes from the DMSOR family, in spite of having its active site molybdenum ion coordinated by two molecules of the pyranopterin cofactor (Figure 1), are also suggested to follow the same general OAT mechanism (Figure 2), as illustrated, for example, by bacterial DMSOR itself (Figure 3d) [57][58][59][60][61][62][63][64][65][66][67][57,58,59,60,61,62,63,64,65,66,67] or bacterial NaRs [64][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][64,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81]. Bacterial nitrite oxidoreductases, that catalyse the oxidation of nitrite to nitrate (NaR reverse reaction), also follow a typical OAT-A mechanism [82][83][84][85][86][87][88][89][90][91][92][82,83,84,85,86,87,88,89,90,91,92]. Noteworthy, to date, no parallel molybdenum-dependent nitrite reductase has been identified.
2. Context—II: The Nitric Oxide Side
Nitric oxide radical (∙NO, herein abbreviated as NO) is a signalling molecule involved in several physiological processes and, consequently, its deficit or excess is associated with many pathological conditions. In humans, NO is produced, in a tightly regulated manner, by three isoforms of NO synthase (NOS; neuronal, endothelial and inducible NOS), using L-arginine and dioxygen as the source of the NO nitrogen and oxygen atoms, respectively (Equation in Figure 4) [93][94][95][96][93,94,95,96]. The “NO signal” is transmitted intra- and extracellularly mainly through post-translational modifications of transition metal centres and thiols. The targets are mostly labile [4Fe-4S] centres and hemes (as is the case of the well-known activation of guanylate cyclase) and cysteine residues, that are converted into the respective nitrosyl (–metal-N=O) and nitrosothiol (-S-N=O) derivates. In addition, to limit the NO toxicity and ensure the fast “on/off signal” response, the NO life time is controlled through its rapid oxidation to nitrate (by reaction with oxy-hemoglobin and oxy-myoglobin [97][98][99][100][101][102][103][104][97,98,99,100,101,102,103,104]) and also to nitrite (by ceruloplasmin [105], cytochrome c oxidase [98][106][107][98,106,107] or dioxygen [108][109][110][108,109,110]) (Figure 4).
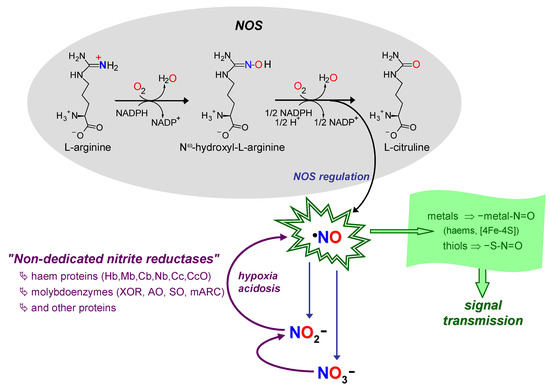
Figure 4. Schematic summary of NO formation–function–extinction. NO formation through NOS and nitrate and nitrite-dependent pathways (black arrows/gray shadowed area and violet arrows, respectively). NO consumption in signalling/function (arrow and shadowed area in green). NO extinction in reactions to limit its toxicity and control the signal specificity (oxidation to nitrate and nitrite; indigo arrows). Hb, haemoglobin; Mb, myoglobin; Cb, cytoglobin; Nb, neuroglobin; Cc, cytochrome c; CcO, Cc oxidase. See text for details.
This NO metabolic flux, formation–function–extinction (Figure 4), where nitrate and nitrite are considered end-products, with no physiological function, was firmly established by the end of the 20th century, when new ideas emerged. It began to become clear that nitrite can be reduced back to NO under anoxic conditions (Equation (9)) and exert a cytoprotective role during in vivo ischaemia and other pathological conditions [111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146]. In accordance, nitrite began to stop being seen as a useless end-metabolite, to become a “storage form” of NO. This new, alternative, NO source would be key to maintain the NO formation and ensure cell functioning under conditions of hypoxia/anoxia, precisely when the dioxygen-dependent NOS activity is impaired (Equation in Figure 4) and a “rescue” pathway is needed.
ONO− + 1e− + 2H+ → ∙NO + H2O
At the same time, numerous studies began to be carried out to identify the pathway(s) responsible for nitrite reduction in mammals (humans), and, later, also in higher plants. The quest did not lead to the identification of any mammalian NO-forming nitrite reductase. Instead, the mammalian nitrite reduction activity was assigned to (already well-known) proteins that are present in cells to perform other (well-known) functions [146][147][148][149][150][151][152][153][154][155][156][157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175][147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176], including, at first, mainly the haem proteins haemoglobin (Hb) and myoglobin (Mb) and the molybdoenzyme XO (Figure 4). However, like all new ideas, the ability of those and other proteins to use nitrite to form and deliver NO faced severe criticism. Many authors questioned the physiological relevance of the NO fluxes generated from the physiologically available (very low) nitrite and called attention to the inhibition by other metabolites. For the “haem community”, the hurdle was the NO delivery, because of the (for long) known ability of haems to easily bind NO and hardly dissociate it. With XO, the main problem was its ability to form NO. The reaction feasibility was controversial, because XO is a well-established OAT-I enzyme (Equation (6)), rather than the OAT-A catalyst needed to reduce nitrite to NO (Equation (9)), and dioxygen was believed to completely inhibit the reaction.