1. Anatomical Overview of Palatogenesis
The palate is divided into two main parts: the hard palate (the anterior bony part) and the soft palate (the posterior muscular part). It separates oral and nasal cavities. During embryogenesis, palatal shelves gradually elevate and come together at the midline, forming the hard palate. The soft palate is formed by soft tissues located posterior to the hard palate, separating the nasal and oral cavities during speech and swallowing. Fusion of palatal shelves begins at their posterior edge and progresses anteriorly
[1][10].
Facial development in humans starts around the fourth week of gestation. The five facial primordia consist of the frontonasal prominence, two mandibular prominences, and two maxillary prominences. Facial development takes place between the fifth and twelfth weeks during embryogenesis. The frontonasal prominence is divided into lateral and medial nasal processes by formation of nasal pits, which subsequently fuse to form the nostril
[1][2][5,10]. Palatal shelves comprise cranial neural crest cells derived from mesenchyme and oral epithelium. Face development requires coordination of a series of formal events, including cell growth, migration, differentiation, and death
[3][11].
The upper lip, philtrum, and primary palate are formed by the union of medial nasal processes and maxillary processes
[4][12]. Their disruptions during the fusion process can lead to cleft lip and/or palate formation. Prospective secondary palates are presented from the oral side of maxillary processes. The secondary palate develops as paired protrusions that grow vertically with the growing tongue and reorientate to a horizontal position across the dorsal portion of the tongue in a process known as palatal shelf elevation. Palatal shelves expand toward the midline, resulting in contact and fusion at this location. The perfect fusion of palatal shelves on each side involves formation of a midline epithelial seam and its disappearance to fill mesenchymal cells. Next, the secondary palate undergoes fusion at its anterior aspect with the primary palate and at its anterodorsal area with the nasal septum, which derives from medial nasal processes at the same period. Finally, the intact roof of the oral cavity is developed, and the oral cavity and nasal cavity are separated. Failure of palatal shelf elevation, contact, and adhesion causes secondary cleft palate. In humans, the development of palate begins at the sixth week of gestation. It is fully accomplished by the twelfth week
[1][10]. In mice, palatal growth is detected at embryonic day (E) 11.5, with palatal fusion being completed by E17
[3][4][5][11,12,13]. Palatogenesis depends on the precise temporal–spatial control of genetic components such as growth factors and signaling molecules for proper development. Several factors including maternal smoking or substance abuse and exposure to environmental toxins can affect palate development. Orofacial clefts result from disruptions of normal biomolecular processes of craniofacial development.
2. Classification of Cleft Lip and Palate in Human
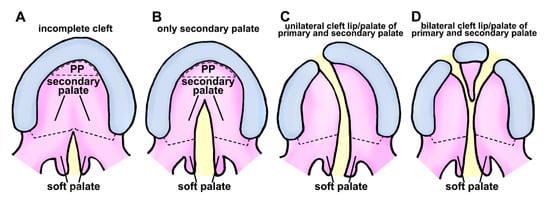
In human, cleft lip and palate are common congenital disabilities that can affect the structure of the face. There are several classifications for describing clefts of the palate and lip. Cleft lip is a congenital condition that occurs when tissues of the upper lip do not fuse together properly during human embryonic development. This can result in a gap or opening in the lip, which can range in size and location. In addition, cleft lip can occur on one side of the lip (unilateral cleft lip) or on both sides of the lip (bilateral cleft lip) (Figure 1).
Figure 1. Schematic representation of cleft palate/lip classification in human. (A) Class I is characterized by an inadequate separation in the palate region, affecting only the soft tissue. (B) Class II clefts impact the secondary palate, consisting of hard and soft palate components. (C) Unilateral separation of primary and secondary palate components characterizes class III. (D) Class IV clefts are characterized by a complete bilateral separation affecting primary and secondary palates. Black dotted semicircular-shape, primary palate (PP); black dotted line, the boundary of the hard palate and soft palate; yellow area represents clefting region.
Unilateral cleft lip is further classified based on the extent of the cleft and location of the cleft within the lip. Unilateral cleft lip of human has three subtypes:
Incomplete cleft lip: This type of cleft lip is characterized by a gap or opening in the lip that is smaller than a complete cleft lip. The location and size of the cleft can vary.
Complete cleft lip: This type of cleft lip involves the entire width of the upper lip, extending from the base of the nose (boundary between the lip and the surrounding skin).
Median cleft lip: This type of cleft lip is a rare type of unilateral cleft lip that occurs in the center of the upper lip, dividing the lip into two separate halves.
Bilateral cleft lip is a type of cleft lip that involves both sides of the upper lip. This type of cleft lip is less common than unilateral cleft lip. It can be more challenging to treat due to the extent of deformity.
Cleft palate is an aberration that arises when the roof of the mouth fails to fuse correctly during human embryonic development. This can result in an opening in the roof of the mouth, which can range in size and location. Cleft palate can occur as a complete cleft (involving both the hard palate and the soft palate) or an incomplete cleft (involving either the hard palate or the soft palate). Unilateral cleft palate has three subtypes (Figure 1):
Complete cleft palate—involves both hard and soft palates.
Incomplete cleft palate—involves either the hard palate or the soft palate.
Submucous cleft palate—involves a small opening in the soft palate, with mucous membrane remaining intact.
Submucous cleft palate is a type of cleft palate that results from a small opening in the soft palate with an intact mucous membrane. This makes it more challenging to diagnose than other cleft palate forms. Consequently, diagnosis might be delayed until speech or hearing issues arise. Cleft lip and palate often occur together.
However, some individuals might have an asymmetric cleft affecting one side of the lip/palate or both sides in opposite configurations, which could impact classification and management
[6][7][14,15]. Evidence involving mouse models specifically for asymmetry in orofacial clefts is limited; however, the formation of orofacial structures can be regulated not only by genetic factors but also by epigenetics and environmental factors. Despite substantial advancements in understanding the genetic etiology of orofacial clefts and accelerated identification of candidate causal mutations through technological and bioinformatic progress, clinical care and prevention strategies remain largely unaffected. This is primarily due to the limited comprehension of the cellular, molecular, and developmental processes underlying cleft pathogenesis
[8][9][10][2,16,17]. Therefore, elucidating the causes through various mouse models is crucial for the development of treatments.
3. Morphological and Molecular Control of Palatal Shelf Growth and Patterning
Lip closure and palatal fusion during human gestation occur at the sixth and twelfth weeks
[4][10][12,17], respectively, necessitating the use of animal models to study normal and abnormal craniofacial development
[3][4][5][8][11][12][2,11,12,13,18,19]. The mouse serves as the primary model organism for investigating orofacial cleft pathogenesis, due to its genetic homology with humans, similar embryonic facial and palate development processes, and the availability of mouse strains with spontaneous or engineered mutations causing cleft lip and/or cleft palate phenotypes
[4][12][13][12,19,20].
Palatal shelves are composed mostly of neural crest-derived mesenchyme
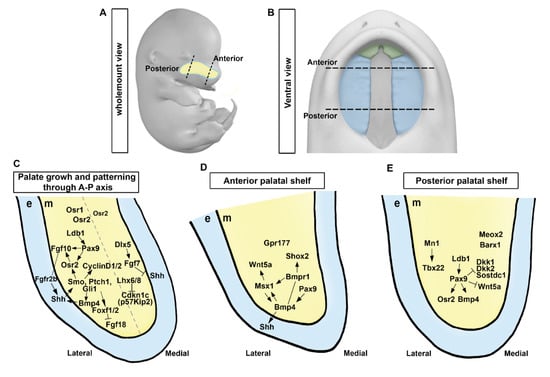
[14][21]. They are bordered by a thin layer of oral epithelium with a unique anterior–posterior (A–P) axis (
Figure 2A,B). At E11.5, the palate is in the early stage of development. At this stage of development, the formation of palatal shelves and the frontonasal process have not yet begun, although neural crest cells that will form palatal shelves have already initiated migration to their proper location. The first signs of the maxillary process can also be seen at this stage. In E12.5, the frontonasal process and palatal shelves have not yet started to lengthen. Around E13.5, palatal shelves, which are two vertically oriented plates of tissue located in the roof of the mouth, begin to elevate and to move towards each other. The frontonasal process located at the front of the head contributes to the formation of the face and elongates to aid in outgrowth
[3][5][15][11,13,22]. The elevating palatal shelves eventually come into contact and fuse in the midline. This process typically occurs between E13.5 and E15.5
[3][11]. Recent studies have shown that palatal shelf growth is regulated by reciprocal epithelial–mesenchymal interactions involving molecular mechanisms along the A–P axis
[16][17][18][19][20][7,8,9,23,24].
Figure 2. Molecular regulation and signaling circuits in palatal shelf growth and patterning. (A,B) A schematic diagram showing the wholemount view of the embryo and the ventral view of the developing palate at E13.5. (C) Signaling pathways governing palatal shelf growth and patterning along the anterior-posterior axis. (D,E) Regulation of growth in anterior and posterior regions of the palatal shelf involves specific molecular pathways, respectively. e, epithelium; m, mesenchyme. Black dotted lines, the anterior and posterior regions in the developing palate at E13.5; Light blue dotted line, developing palate; Gray dotted line, the hypothetical margin of the lateral and medial side in the palatal shelf. Arrows represent inductive relationships. Solid lines represent direct physical interaction. Blunt arrows indicate inhibition.
Previous studies have identified essential components of signaling pathways for palatal shelf outgrowth. Several signaling molecules, including sonic hedgehog (Shh), wingless and Int-1 (Wnt), fibroblast growth factor (Fgf), and bone morphogenetic protein (BMP), can regulate palatal shelf outgrowth, with Shh playing a key role in palatal shelf outgrowth
[8][17][18][2,8,9]. Inactivation of Shh in the epithelium or mesenchyme-specific inactivation of
Smoothened (
Smo) can impair palatal cell proliferation and outgrowth, indicating that Shh signaling is vital for mitogenic response of palatal cells
[21][25]. Evidence for the role of SHH signaling in facial growth has been demonstrated by manipulating Hhat, which encodes an acyltransferase involved in modifying Hh proteins, and Ptch1
[22][26]. Mice with concurrent
Hhat and
Ptch1 mutations displayed SHH gradient disruptions during frontonasal process development, resulting in medial and lateral nasal process hypoplasia and ultimately causing cleft lip and remaining midline epithelial seam
[22][26]. Primary cilia are tiny hair-like projections that extend from the surface of many tissues in the body
[23][27]. They play an important role in transmitting Shh signaling, as evidenced by reduced expression of
forkhead box F1 (
Foxf1) in the palatal mesenchyme
[24][25][26][28,29,30], suggesting that primary cilia might function as downstream effectors of Shh signaling.
Fgf10−/− can lead to cleft palate with impaired palatal shelf outgrowth
[27][31]. Although
Fgf10 expression is in the mesenchyme, its receptor
Fgfr2b is detected in the overlying epithelium. Fgfr2 is required in the epithelium since an epithelial-specific deletion of
Fgfr2 also leads to cleft palate
[28][32]. Odd-skipped related 2 (
Osr2) is decreased in the palatal mesenchyme when Shh signaling is deleted
[29][33].
Osr2 expression in palatal mesenchymal cells depends on the function of
Pax9 [30][31][34,35]. Embryos with
Osr2−/−;
Pax9−/− exhibit cleft palate and reduction of
Fgf10 expression in the palatal mesenchyme
[30][31][34,35]. Indeed, Shh is intensively reduced in the epithelium of
Fgf10−/− and
Fgfr2b−/− embryo, indicating that decreased palatal mesenchymal proliferation observed in these mutants might be due to decreased Shh expression in the epithelium
[27][31]. Shh signaling acts in concert with FGF signaling by a positive feedback loop to control palatal epithelial and mesenchymal proliferation
[27][29][31,33] (
Figure 2C). Together, these two signaling pathways and transcription factors ensure proper formation of the palate and establishment of oral and nasal cavities by activating a mesenchymal signal (
Figure 2C–E).
Fgf10 plays a role in maintaining Shh expression in the palatal epithelium
[27][31], whereas
Fgf7, a closely related member of the FGF family, has the opposite effect by suppressing
Shh expression in the palatal epithelium
[32][36].
Fgf7 expression in the medial palatal mesenchyme is regulated by
Dlx5, which restricts
Shh expression to the lateral palatal epithelium
[32][36]. Emerging research on genetic and explant culture investigations has unveiled crucial molecular mechanisms underlying palatal shelf formation, which involve the intricate interplay among Shh, Foxf1/2, and Fgf18 within a complex regulatory network
[20][24]. Ablation of both
Foxf1 and
Foxf2 in the neural crest of mouse embryos leads to defective development of palatal outgrowths and abnormal
Fgf18 expression in the palatal mesenchyme
[20][24]. In addition,
Foxf2−/− mouse embryos display restricted palatal shelf growth. Such defective growth is characterized by mislocalized, increased expression of
Fgf18 in specific regions of the palatal mesenchyme, and loss of
Shh expression in complementary areas of the palatal epithelium
[20][24]. Several transcription factors and unique FGF ligands, all of which are controlled by the Shh signaling pathway, coordinate many subnetworks that govern the growth and patterning of the palatal shelf
[18][9] (
Figure 2C).
In addition, LIM homeobox 6 (
Lhx6) and LIM homeobox 8 (
Lhx8) can regulate maxillary arch and palatal mesenchyme proliferation by suppressing expression of genes encoding FOX family transcription factors and
Cdkn1c (also known as
p57Kip2), a cell cycle inhibitor
[33][37]. While the
Shh-Foxf1/2-Fgf18-Shh molecular circuit has been recently found to be engaged in early development of the palate
[20][24], it remains unknown whether
Lhx6/8 modulates Shh and FGF signaling network during formation of the palatal shelf (
Figure 2C). Furthermore, transforming growth factor-β (TGF-β) signaling influences Shh signaling in the palatal mesenchyme by modulating lipid metabolism
[34][38].
Shh and Bmp signaling pathways have been discovered to be able to interact with each other (
Figure 2C,D). In the palatal mesenchyme, deletion of
Smo leads to overexpression of
Bmp4 and downregulation of
Bmp2 [29][33]. Shh signaling can stimulate the expression of
Bmp2, as confirmed by induction of
Bmp2 expression in palatal explant culture with Shh-containing beads
[35][39]. Exogenous Bmp2 can also promote cell proliferation in the palatal mesenchyme
[35][39]. Although complete inactivation of
Bmp4 was fatal during early stages of embryonic development, targeted ablation of
Bmp4 function solely in the maxillary mesenchyme and oral epithelium resulted in the development of cleft lip. At the same time, a no concomitant defect in the secondary palate was detected
[36][40]. Palatal mesenchyme-specific overexpression of BMP antagonist Noggin in mice resulted in retarded palatal growth and cleft palate
[37][41], further supporting the requirement for of BMP signaling for normal palatogenesis.
Previous studies identified that type I Bmp receptor
Bmpr1a is a mediator of Bmp signaling in palate formation
[36][40]. Deletion of
Bmpr1a in the maxillary mesenchyme and oral epithelium of mice (in
Nestin-Cre; Bmpr1af/− mice) resulted in cleft lip and palate
[36][40], while epithelial-specific loss of
Bmpr1a (in
K14-Cre; Bmpr1af/−) did not cause cleft palate
[38][42]. These findings suggest that Bmpr1a signaling in the palatal mesenchyme, rather than oral epithelium, is necessary for proper palatogenesis (
Figure 2C). On the other hand, conditional deletion of
Bmpr1a in the neural crest (in
Wnt1-Cre; Bmpr1af/− mouse embryos) and its derivatives can cause severe retardation in the anterior region of palatal shelves accompanied by various craniofacial defects
[39][43]. Inactivation of
Bmpr1a within the palatal mesenchyme was achieved using
Osr2-IresCre; Bmpr1af/f mice, which resulted in an anteriorly restricted cleft palate and decreased cell proliferation in the anterior palatal mesenchyme. In addition, loss of
Bmpr1a in
Osr2-IresCre; Bmpr1af/f reduced cell proliferation and
Shh expression in both primary and secondary palates, indicating that BMP–SHH interactions could regulate palate outgrowth
[40][44]. Furthermore, loss of BMP antagonist Noggin caused cleft palate, with aberrant apoptosis in the palatal epithelium and reduced mesenchymal cell proliferation
[41][45]. This demonstrates that strict regulation of BMP signaling is required for normal palate development (
Figure 2C).
WNT signaling is vital in
Pax9-mediated secondary palate development
[18][19][42][43][44][9,23,46,47,48]. In
Pax9−/− mice, posterior palate levels of
Axin2 and activated
β-catenin—direct targets of canonical WNT signaling—were diminished, coinciding with increased WNT antagonist
Dkk2 expression
[18][42][9,46]. Pharmacological inhibition of
DKK activity using small-molecule agonists IIIc3a
[18][42][9,46] or WAY-262611
[44][48] partially rescued palate morphology and fusion, leaving a minor cleft. Despite decreased
Sostdc1 and
Bmp4 expression in
Pax9−/− mice, genetic inactivation of Sostdc1 sufficiently restored canonical WNT signaling in the palatal mesenchyme and rescued cleft palate
[18][42][9,46].
In other developmental contexts, EDA/EDAR signaling is downstream of WNT signaling, and
Eda expression is reduced in
Pax9−/− mice
[43][47]. Although EDA/EDAR signaling is not essential for palate formation
[45][49], in utero stimulation with an EDAR agonist rescued cleft palate in
Pax9−/− mice
[43][47]. Treated mice exhibited disorganized rugae and unaltered
Bmp4,
Msx1,
Fgf10, and
Osr2 expression, while WNT pathway component analysis was not performed. These studies imply that
Pax9 influences WNT signaling through modulation of WNT antagonists in the palatal mesenchyme, although further investigation is needed to understand the transcriptional regulation of WNT target genes.
4. Regionalization of Anterior and Posterior Palatal Outgrowth
Developing palatal shelves exhibit distinct molecular and morphological differences along the A–P axis, with specific pathways functioning in anterior and posterior compartments
[46][47][48][50,51,52] (
Figure 2C–E). Developing palatal shelves exhibit differential gene expression along the A–P axis, with various transcription factors including BarH-like homeobox 1 (
Barx1), meningioma 1 (
Mn1), Msh homeobox 1 (
Msx1), mesenchyme homeobox 2 (
Meox2), short stature homeobox 2 (
Shox2), and T-box transcription factor 22 (
Tbx22) being expressed in distinct regions
[35][46][47][48][49][50][39,50,51,52,53,54]. For instance,
Msx1 and
Shox2 are expressed in the anterior palate, while
Meox2 and
Tbx22 are expressed in the posterior region, with the boundary aligned with the first-formed palatal rugae
[35][46][47][48][49][50][39,50,51,52,53,54]. Transcription factors
Msx1 and
Shox2 are crucial for stimulating cellular proliferation in the anterior palatal mesenchyme where they are mainly expressed
[35][50][39,54].
Msx1 also plays a crucial role in maintaining Shh expression in the anterior palatal epithelium by regulating the expression of
Bmp4 in the anterior palatal mesenchyme
[35][39].
Barx1 and
Mn1 mRNAs are expressed mainly in the posterior palate. However, their expression domains also partially overlap with the anterior half of palatal shelves
[48][51][52,55]. Mice with disrupted
Msx1 or
Mn1 gene expression exhibit a complete cleft palate while
Msx1−/− mice exhibit proliferation defects only in the anterior region and
Mn1−/− mice exhibit growth deficits confined to middle and posterior regions of the palatal shelves
[35][51][39,55]. Conversely,
Shox2−/− animals show a cleft limited to the anterior palate, whereas the posterior palate shows normal fusion, indicating a particular function for
Shox2 in anterior palatal expansion
[50][54] (
Figure 2D).
Tbx22−/− mice displayed cleft palate, ranging from full cleft palate owing to shortened palatal shelves to the submucous cleft palate with normal palatal shelf elevation and fusion
[52][56].
Tbx22 mRNA expression is significantly reduced in
Mn1−/− mice and
Mn1 is capable of stimulating
Tbx22 expression in cell culture assays, indicating that
Tbx22 is downstream of
Mn1 in regulating posterior palatal outgrowth
[51][55] (
Figure 2E).
Proper regulation of
Msx1 and
Shox2 expression in the anterior palatal mesenchyme depends on Bmp signaling, as demonstrated by marked decreases of expression levels of both genes in the anterior palate of mice with
Wnt1-Cre; Bmpr1af/− genotype
[39][43]. Remarkably, in palatal explant cultures,
Msx1 expression was induced only in the anterior palatal mesenchyme by adding Bmp4-soaked beads
[46][50], whereas
Shox2 mRNA expression in palatal mesenchyme explants could not be induced by exogenous Bmp4
[50][54]. However, anterior palatal epithelium induced ectopic
Shox2 mRNA expression in the posterior palatal mesenchyme
[50][54]. These results reveal intrinsic distinctions between the epithelium and mesenchyme along the A–P axis (
Figure 2D).
Canonical Wnt signaling in the developing palatal mesenchyme is restricted to the anterior area, as identified by the
BATGAL transgenic reporter and canonical Wnt signaling is dependent on
Gpr177-mediated Wnt secretion by neural crest-derived mesenchyme
[53][57]. Furthermore,
Wnt5a is highly expressed in the anterior palatal mesenchyme. It governs palatal mesenchyme migration and palatal shelf elongation
[54][58]. Msx1 can promote transcriptional activation of
Wnt5a in the anterior palatal mesenchyme via an enhancer that originates from a transposable element
[55][59]. On the other hand, LIM domain-containing transcription factors require cofactor
Ldb1 for proper palatal shelf growth and A–P patterning. In particular, neural crest-specific inactivation of
Ldb1 can induce ectopic
Wnt5a in the posterior palatal mesenchyme
[56][60], indicating that certain LIM domain-containing transcription factors might play a significant role in the growth of palatal shelf and A–P patterning.
5. Patterning along the Mediolateral Axis
Patterning along the mediolateral axis of the palate involves establishment of distinct gene expression domains that are necessary for proper palatal development. This patterning along the mediolateral axis in palate development is significant because it builds an intricate network of signaling channels and gene domains required for optimal palatal growth and fusion (
Figure 2C). During vertical outgrowth of developing palatal shelves, morphological and molecular heterogeneity is visible along the mediolateral axis; the oral side aligned with the lateral side after elevation of the palatal shelf. In addition, around E12, the lateral side of the developing palatal shelves commences production of palatal rugae, concurrent with restriction of
Shh expression to the lateral palatal epithelium
[26][30].
Osr1 and
Osr2 are zinc-finger transcription factor encoding genes that demonstrate graded expression along the mediolateral axis of the developing palatal mesenchyme
[30][34]. At E13.5,
Osr1 mRNA is limited to the lateral side. However,
Osr2 displays graded expression which is the strongest in the lateral mesenchyme. It progressively decreases toward the medial mesenchyme. Deletion of
Osr2 results in formation of cleft palate, which is accompanied by decreased cell proliferation on the medial side of the developing palatal shelves as well as the disruption of mediolateral patterning
[57][61]. Partial functional redundancy of
Osr2 and
Osr1 is likely responsible for the necessity of
Osr2 in mediating cell proliferation, particularly on the medial side since cleft palate could be repaired in
Osr2-deficient mice by replacing
Osr2 coding sequence with an
Osr1 cDNA
[57][61]. In
Osr2−/− mice, transcriptional profiling and expression analysis demonstrated an increase in osteogenesis-related genes, such as
Mef2c,
Sox6,
Sp7, and various BMP ligands (
Bmp3,
Bmp5, and
Bmp7)
[58][62]. In addition, class-3 semaphorins (
Sema3a,
Sema3d, and
Sema3e) were found to be ectopically expressed and identified as direct targets of
Osr2 (specifically
Sema3a and
Sema3d). These findings indicate that
Osr2 has a crucial role in controlling mesenchymal cell proliferation and fate by inhibiting premature osteogenesis and abnormal semaphorin expression. Nevertheless, more research is required to comprehend the role of semaphorins in palate development
[58][62].
A pathway involving transcription factor
distal-less homeobox 5 (
Dlx5) can regulate mediolateral patterning and palatal expansion. In the medial mesenchyme of the palatal shelf,
Dlx5 is co-expressed with
Fgf7. Fgf7 expression is markedly decreased in this region in
Dlx5-deficient mutant palatal shelves
[32][36]. In
Dlx5-deficient mutant embryo, expansion of
Shh expression into the medial palatal epithelium might be attributed to loss of
Fgf7, as demonstrated by the ability of exogenous Fgf7 to inhibit
Shh expression in palatal explant cultures
[32][36]. While palate shelves are elevated and fused in
Dlx5-deficient individuals, the oral part of the palate is dramatically enlarged, and a deformed soft palate is visible. Intriguingly, while
Msx1-deficient mice showed decreased
Shh expression in the anterior palate, compound mutant embryos lacking both
Dlx5 and
Msx1 displayed Shh expression in the medial palatal epithelium, which was able to compensate for cell proliferation defects associated with
Msx1 loss-of-function
[32][36]. Collectively, these findings identify a novel pathway involving
Dlx5 and
Fgf7 in regulating mediolateral patterning and palate growth. However, given that mice lacking
Fgf7 did not show any apparent palatal defects
[59][63], another signaling molecule might function downstream of
Dlx5, possibly in conjunction with
Fgf7, to modulate
Shh expression in the palatal epithelium (
Figure 2C).
6. Genetic Network Controlling Palatal Shelf Adhesion and Fusion
As the palatal shelf grows, maxillary and mandibular processes also grow. This enables the tongue to move downward and forward, which is necessary for elevating the palatal shelf. Upon their elevation, palatal shelves contact with each other at the midline and fuse
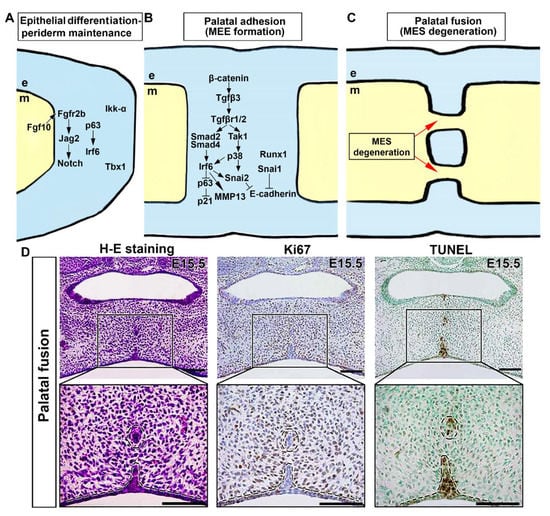
[60][64]. A complex network of signaling pathways regulates the adhesion and fusion of the developing palatal shelves. Many important signaling pathways and genes are involved in epithelial differentiation of the palate. To establish mesenchymal continuity throughout the fused palate, it is necessary to eliminate the intervening epithelium between adjoining palatal shelves, which is referred to as midline epithelial seam (MES) (
Figure 3). A cleft palate may result from disrupting spatial and temporal control of midline edge epithelium (MEE) differentiation, adhesion competence, and disappearance of MES.
Figure 3. Molecular and cellular processes underlying adhesion and fusion of the palate. (A) Diverse intracellular pathways and transcription factors in the medial edge epithelium (MEE) and the periderm above instigate cell cycle exit, disturb epithelial adhesion, and degrade extracellular matrix (ECM). (B) Molecular control of palatal epithelial differentiation. (C) Morphological changes in midline epithelial seam (MES) during palatal fusion. (D) Histologic, cell proliferation, and cell death analysis of MES degeneration in the palatal fusion region at E15.5. Histological analysis indicates degeneration of MES during palatal fusion, with concurrent absence of cell proliferation and cell death in the remaining MES. Black, magnification of palatal fusion region. Black dotted circle, remaining MES during palatal fusion. Black dotted line, the margin of palatal epithelium and mesenchyme. Arrows represent inductive relationships. Solid lines represent direct physical interaction. Blunt arrows indicate inhibition. e, epithelium; m, mesenchyme. Scale bar: 100 μm.
Animals with malfunctioning
Jag2,
Fgf10,
Irf6, and
Grhl3 genes have a cleft palate phenotype and aberrant adhesion or fusion of palatal shelves with the mandible and/or tongue
[61][62][63][64][65][65,66,67,68,69]. Absence of
Jag2 Notch ligand can cause cleft palate in
Jag2zΔDSL/ΔDSL mice primarily due to abnormal adhesion of palatal shelves to the tongue
[66][70].
Jag2 is expressed in the oral epithelium. It is responsible for maintaining periderm cells, which are believed to regulate fusion competence
[62][67][66,71]. Moreover, palate–tongue fusion, although not severe, and decreased expression of
Jag2 in the palatal epithelium have been found in
Fgf10−/− embryos, indicating that Fgf10 signaling can regulate palatal epithelial development upstream of Jag2-Notch signaling
[61][65]. Mice lacking functional interferon regulatory factor 6 (
Irf6) due to homozygous null mutations or an R84C point mutation show a hyperproliferative epidermis that does not differentiate, resulting in a range of developmental abnormalities, including cleft palate and inappropriate oral adhesions
[63][65][67,69]. Irf6 can regulate periderm differentiation in collaboration with
Jag2, as evidenced by the development of palate–tongue fusion, oral adhesions, and cleft palate in compound
Irf6R84C/+; Jag2ΔDSL/+ mice
[68][72]. This phenotype resembles that seen in mice with homozygous
Irf6 or
Jag2 alleles, emphasizing the significance of these genes in palatal development. The expression of each gene was unaffected in the reciprocal individual mutant, indicating that Irf6 does not directly regulate
Jag2 expression
[65][69].
It has been found that
p63 transcription factor-deficient mice exhibit a cleft palate and a thin, undifferentiated epidermis
[69][70][73,74], with reduced Irf6 expression in the palatal epithelium
[71][75]. In addition, heterozygous mutant mice, compound
p63+/–; Irf6R84C/+, exhibit a failure in palatal shelf fusion associated with improper preservation of periderm cells.
p63 can exert a positive regulatory effect on the expression of
Jag2 and
Fgfr2 in various other cell types
[72][73][76,77]. Although the relationship between p63, Jag2-Notch, and Fgf10-Fgfr2b signaling pathways in palatal epithelial differentiation is not yet fully understood, previous studies suggest that
p63 might positively regulate
Jag2 and
Fgfr2 expression in other cell types. In addition, a lack of
Ikk-α or
Tbx1 in mouse embryos results in aberrant oral adhesions between the tongue and palatal shelves
[74][75][78,79]. These findings suggest that palatal epithelial differentiation is regulated by a genetic network involving Irf6, Jag2, p63, Ikk-α, Tbx1, and Fgf10-Fgfr2b signaling pathways (
Figure 3A).
Although periderm is essential to prevent abnormal oral epithelial adhesions, it must be removed from the medial edge of the palatal shelf to initiate fusion. Precise mechanisms responsible for controlling periderm removal have not yet been fully elucidated. The MES is the structure that separates palatal shelves prior to fusion. There are three main hypotheses explaining how the MES disappears. One hypothesis is that the MES disappears due to epithelial-to-mesenchymal transition (EMT), which could allow the intervening epithelium to be incorporated into the mesenchyme of the intact palate. For example, genetic lineage tracing using epithelial-restricted Cre-expressing transgenic lines paired with the ROSA26R (R26R) reporter line has been used to track the destiny of MES cells in vivo
[76][77][80,81]. In one study,
lacZ expression was specifically and irreversibly activated within the epithelium of
ShhGFPCre or
K14-Cre mice crossed with R26R reporter mice. Subsequent examination of β-galactosidase staining during and following MES removal allowed the fate of MEE cells to be followed to determine whether they contributed to the mesenchyme (i.e., if they underwent EMT)
[76][80]. This approach did not detect
lacZ-expressing mesenchymal cells, concluding that EMT was not a significant contributor to the regression of the MES
[76][80]. However, a third group found mesenchymal β-galactosidase activity during and soon before regression of the MES in
K14-Cre; R26R embryos
[78][82]. The authors suggested that the disagreement might be due to variations in Cre expression levels and/or patterns in the palatal epithelium of several
K14-Cre transgenic mice lines utilized.
Apoptosis has been shown to play a significant role in MES dissolution to obtain mesenchymal confluency. Cell proliferation is rarely observed at that location (
Figure 3D). Several studies have shown that many MES cells are TUNEL positive and active caspase 3 positive during palatal fusion
[76][79][80][81][80,83,84,85]. A new genetic research has studied the influence of the
Apaf1 gene, which encodes an essential component of caspase 3-mediated apoptosis, on palatal fusion and found that
Apaf1 deficiency does not impair palate fusion or MES dissolution
[82][86]. This observation contrasts with a previous report indicating that palatal shelves could make contact but fail to fuse in
Apaf1-deficient embryos
[79][83]. However, that study did not perform a thorough evaluation of the secondary palate. Although apoptosis plays a substantial role in MES disintegration, further study is required to clarify the participation of other cellular processes, namely the fusion mechanism between the anterior secondary palate and primary and secondary palates. Despite this, the significance of TGF-β signaling in eliminating MES is clear since
Tgf-β3 is solely expressed in the MEE. The lack of
Tgf-β3 in embryonic mice allows palatal shelves to establish improper contact at the midline, resulting in persistence of the MES
[83][84][85][87,88,89].
Contact between type I and type II receptor dimers can activate the Tgf-β signaling pathway, leading to phosphorylation of R-Smads and transcriptional regulation.
Smad2 knockdown in palatal explants can prevent the breakdown of the MES, while
Smad2 transgenic overexpression in the palatal epithelium partly repairs palate fusion in
Tgf-β3 deficient animals
[86][87][90,91]. Nevertheless, epithelial-specific deletion of
Smad4 in
K14-Cre; Smad4f/f mice did not affect palatal shelf fusion, suggesting the involvement of other pathways
[87][88][91,92]. Tgf-β signaling may trigger the p38 MAPK pathway, which is increased in the palatal epithelium undergoing fusion. Tgf-β signaling triggers activation of
Tgf-β activated kinase 1 (
Tak1), which operates separately from the Smad pathway, to initiate activation of the p38 MAPK pathway. Both pathways work redundantly to promote palatal fusion
[3][88][11,92]. Inhibition of
p38 MAPK in
K14-Cre; Smad4f/f palatal explants prevents Tgf-β-dependent expression of the
p21 gene, reducing apoptosis and MES dissolution failure
[88][92]. These findings suggest that
Smad- and
p38 MAPK-dependent mechanisms are functionally redundant during palate fusion. Notable,
Irf6, a vital factor responsible for periderm differentiation, is activated not only in the periderm layer but also in the basal layer of MEE cells before fusion of palatal shelves
[89][93]. Failure of palatal fusion and diminished MEE expression of Irf6 have been observed in
K14-Cre;Tgfβr2f/f mutant embryos
[89][93]. However, overexpression of Irf6 in basal epithelial cells has been found to restore palatal fusion in
K14-Cre;Tgfβr2f/f embryos
[89][93]. The
Irf6 expression functions during periderm differentiation, leading to the downregulation of
p63 and increased
p21 expression in MEE cells. This mechanism is believed to facilitate cell cycle exit and subsequent degeneration of the MEE
[89][90][93,94] (
Figure 3A). Tgf-β3 is crucial for downregulation of
Jag2 in the MEE. Blocking Notch signaling can partially restore fusion between
Tgf-β3-deficient palatal shelves in explant culture
[91][95]. Maintenance of oral periderm integrity depends on Jag2-Notch signaling
[62][66]. Therefore, reduction of
Jag2 expression in the MEE is likely a key mechanism by which
Tgf-β3 disrupts periderm function and facilitates palatal shelf adhesion. A previous study revealed that beta-catenin (Ctnnb1) regulates MES dissolution by controlling
Tgf-β3 expression in the MEE. In epithelial-specific
beta-catenin (
Ctnnb1) disruption experiments, reduction of apoptotic MES cells and loss of
Tgf-β3 expression in the MEE were observed, resulting in cleft palate due to failed palatal shelf fusion
[92][96]. However, given that
beta-catenin (
Ctnnb1) can function as either a component of adherent junctions or in the canonical Wnt signaling pathway
[92][96], the precise mechanism underlying its involvement in this context remains to be elucidated through further investigation.
Several transcription factors play a crucial role in the initiation of palatal fusion (
Figure 3A,B). The Snail family of transcription factors is essential in regulating palatal fusion, as evidenced by the failure of fusion in
Snai1+/–;
Snai2+/– compound mutants, which coincides with a reduction in MES apoptosis
[93][97]. Expression of
Tgfβ-3 was not affected in these mutants
[93][97], but exogenous Tgf-β3 in cultured primary MEE cells was found to induce the expression of
Snai1 via a pathway independent of Smad signaling. These results indicate that these transcription factors could regulate palatal fusion downstream of or in parallel with the Tgf-β3 pathway. Interestingly,
Runx1 is another transcription factor studied in the context of palate development (
Figure 3A,B). Although it is expressed in the MEE throughout the AP axis during palate fusion, its disruption can lead to anterior-restricted failure of palatal shelf fusion and failed fusion with the primary palate. This anterior cleft is associated with a unique region of the MEE that exhibits less TUNEL staining and distinct behavior compared to the rest of the palatal shelf in unpaired palatal culture, suggesting that
Runx1 might play a role in this anterior MEE behavior
[21][25]. In contrast, the
Meox2 transcription factor is required to maintain palatal integrity after successful fusion and dissolution of the MES.
Meox2−/− embryos exhibit a post-fusion split of the posterior palate
[82][86].
Irf6 is required for
Snai2 expression in MEE cells.
Snai2 knockdown slows palatal fusion in explant cultures
[94][98]. Activation of
Ephrin reverse signaling enhances MEE expression of
Snai1 in palatal explant cultures and partly repairs palatal shelf fusion in the presence of
Tgf-β3 function-blocking antibodies
[95][99]. These data imply that ephrin reverse signaling and Tgf-β3 signaling might cooperate to regulate palatal fusion.
Snai1 and
Snai2 are transcription factors that play a role in the epithelial–mesenchymal transition process. They act downstream of Tgf-β3 signaling. Snai family is known to downregulate the expression of
E-cadherin [18][9]. This downregulation may contribute to loosening of the medial edge epithelium (MEE) and periderm cell adhesion, resulting in periderm desquamation.
Tgf-β3 and
Irf6 are crucial for activating the expression of
MMP13 in the palatal MEE, which may contribute to periderm desquamation by breaking down the basement membrane. In addition, carcinoembryonic antigen-related cell adhesion molecule 1 (
CEACAM1) expression is upregulated in MEE periderm cells before palatal fusion and
Ceacam1−/− mouse embryos display a delay in palatal fusion completion
[96][100].
Tgf-β3 expression in palatal MEE remains unaffected in
Ceacam1−/− embryos. It is uncertain if
CEACAM1 functions downstream of
Tgf-β3 to regulate periderm desquamation and/or palatal shelf adhesion. The connection between molecular processes that lead to desquamation and apoptosis and other molecules involved in
Tgf-β3-induced periderm cell apoptosis requires further investigation.
The genetic process of palatal development was described previously, and several studies additionally showed the role of epigenetic factors such as microRNAs that regulate genes in the palatal fusion process
[97][98][101,102]. For example, miR-200b, highly expressed in epithelial cells
[99][103], was found in the MES during palatal fusion and its expression decreased as fusion progressed.
Smad2, essential for
Snai1 induction in Tgf-
β signaling during palate development, was expressed in MEE and MES
[97][98][101,102].
Snai1, crucial for palatal fusion via Tgf-
β signaling, was present in mesenchyme and some MEE cells
[98][102]. In addition, Ectopic miR-200b expression led to Zeb family suppression,
E-cadherin upregulation, and alterations in cell migration and palatal fusion
[97][101]. These findings indicate the critical role of miR-200b in cell migration and palatal fusion during palate development by regulating
Zeb1 and
Zeb2 as a noncoding RNA, while also suggesting a potential interaction with TGF-
β-mediated
Smad2 and
Snai1 signaling pathways in the context of normal palate development.
While the MES ultimately undergoes degeneration, the epithelia situated on the nasal and oral facets of the palate differentiate into pseudo-stratified, ciliated columnar cells and stratified, squamous, keratinizing cells, respectively. Although epithelial differentiation is dictated by the subjacent mesenchyme
[60][64], the molecular determinants governing oral and nasal epithelial cell fate remain elusive. Additionally, the palatal mesenchyme differentiates into osseous and muscular tissues, constituting the hard and soft palate, respectively; recently, reviews have explored the molecular mechanisms delineating these discrete cell fates
[100][101][104,105].