Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Fanny Huang and Version 1 by Qiong Wang.
Cancer cells circumvent immune surveillance via diverse strategies. In accordance, a large number of complex studies of the immune system focusing on tumor cell recognition have revealed new insights and strategies developed, largely through major histocompatibility complexes (MHCs). MHC-I molecules are known as human leukocyte antigens (HLA) and HLA class-I molecules are composed of classical (class-Ia) and the non-classical (class-Ib) components.
- MHC-I
- immune evasion
- major histocompatibility complexes (MHCs)
1. Introduction
In recent decades, cancer immunotherapies like chimeric antigen receptor-engineered T cell (CAR-T), checkpoint blockade of PD-1/PD-L1, etc., are emerging in an endless stream which arrests the attention of physicians, patients and researchers from all over the world for their different, genius and extraordinary efficiency on cancer treatment compared with traditional chemotherapy and radiotherapy. This demonstrates that cancer generation and development are closely bound up with immunological events in the body.
In general, extrinsic pathogens and abnormal intrinsic cells with senescent, malignant transforming, infected, dysfunctional or other phenotypes should be targeted by immune cells and, in consequence, be killed. In specific immune responses to the special antigens, there are major histocompatibility complexes, commonly including MHC-I and MHC-II molecules that play key roles in cell-intrinsic and pathogen-extrinsic antigen targeting and presentation, respectively. All nucleated cells in the body express MHC-I molecule and cellular-intrinsic antigens are thereby processed and presented by MHC-I. For example, an infected cell combines pathogen peptide with MHC-I and presents the complex, or a malignant cell combines cancerous peptide with MHC-I and presents the complex. Those complexes can be recognized by cytotoxic T lymphocytes (CTLs), such as CD8+T cells, and apoptosis of the targeted cell is initiated as a consequence, while extrinsic pathogens, including bacteria, fungi, viruses, chlamydia or mycoplasma, can be recognized and engulfed by antigen-presenting cells (APCs) via endocytosis. Thereafter, extrinsic peptides are presented by APCs through being combined with MHC-II molecules. Usually, MHC-II molecules are identified by CD4+T cells which then play a role as a helper in mediating CD8+T cell activation and inducing B lymphocyte differentiation, and then the latter two initiate cellular immunity and humoral immunity, respectively.
2. MHC-I Reduction and Immune Evasion
In cancer immune surveillance, T lymphocyte-mediated specific immune response plays a crucial role. However, cancer immunity can be blocked through the alteration of the tumor microenvironment, leading to surveillance evasion. Thus far, the related strategies include: decreasing cancer antigens’ immunogenicity and rendering cancer cells less visible, muting APCs, inhibiting naïve T cell stimulation, depressing CTL infiltration into the tumor, suppressing CTLs’ cytotoxic efficacy, etc. [1,2,3][1][2][3]. Clinical cancer immunotherapies may, therefore, suffer from cancer cell-mediated immune invisibility through immune evasion mechanisms, which usually result from the downregulation of MHC-I expression or the failure of antigen presentation (Figure 1). In human cells, MHC-I molecules are known as human leukocyte antigens (HLA) and HLA class-I molecules are composed of classical (class-Ia) and the non-classical (class-Ib) components [4,5,6,7][4][5][6][7]. In fact, HLA-Ia expression defects are always found followed by uncontrolled growth of both primary and metastatic cancers [8[8][9][10],9,10], demonstrating their correlation with high tumor grading and progression, decreased survival and ineffective immunotherapies [11,12,13,14][11][12][13][14]. An MHC-I expression defect could derive from reversible lesions and irreversible lesions. The former are aroused from epigenetic modifications such as IFNγ-induced MHC-I expression [15,16][15][16], while the latter are aroused from less common structural mutations. Through investigating the regulation of MHC-I expression, Kobayashi et al. found that NOD-, LRR- and CARD-containing 5 (NLRC5) notably upregulated MHC-I, β2-microglobulin (β2M) and transporter associated with antigen processing 1 (TAP1), while the latter two as indispensable parts play key roles in the MHC-I antigen presentation pathway [17]. This study also revealed that NLRC5 was induced by IFNγ in a dose-dependent manner, indicating that NLRC5 probably plays a key role in the MHC-I antigen presentation pathway. Subsequently, they also reported in the following studies that NLRC5 expression was lost in varying cancers and the loss was correlated with the loss of CTL activation in several types of cancer [18].
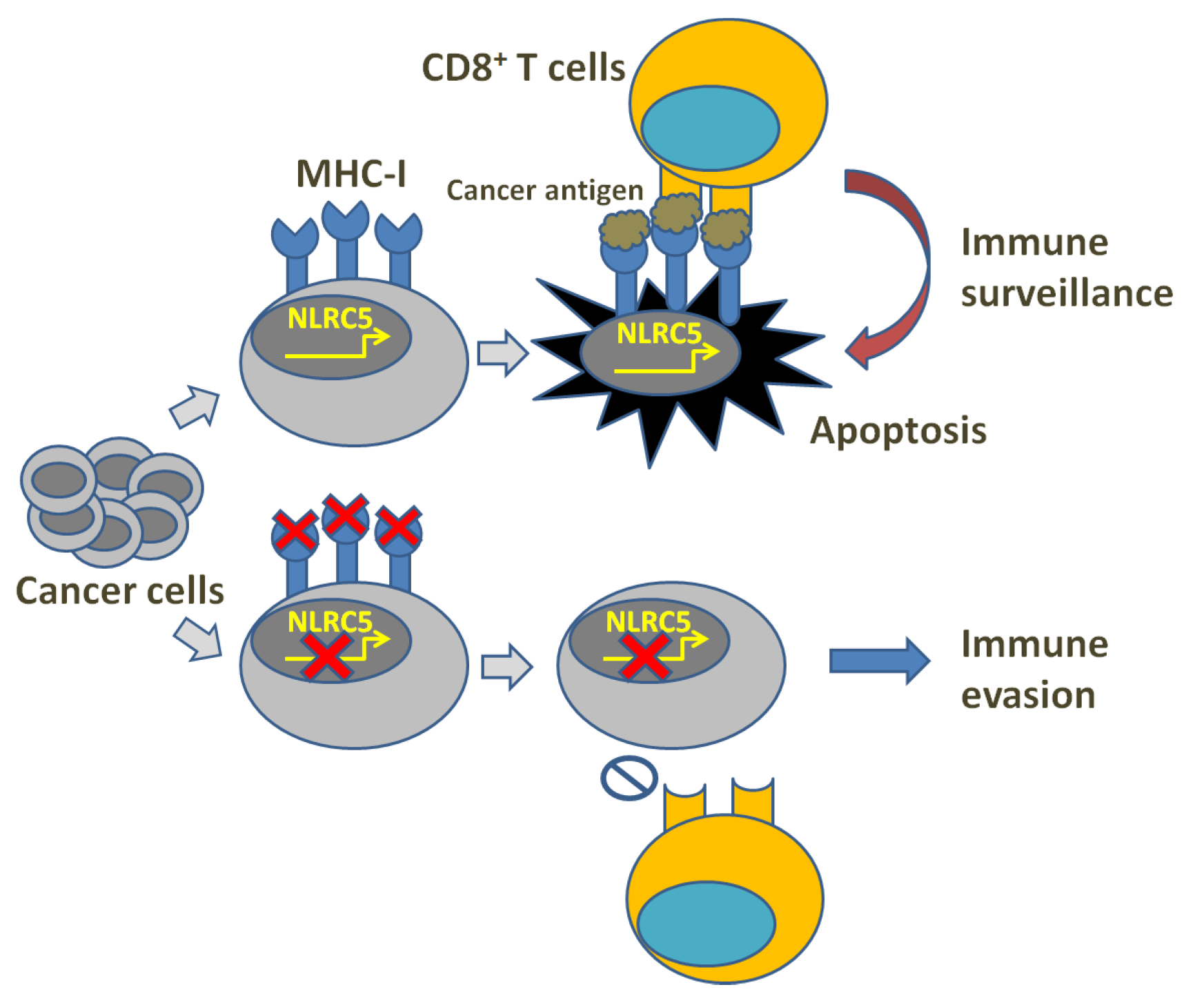
Figure 1. Illustration of MHC-I deficiency and immune evasion.
It seems that NLRC5 expression is not invariable in different tissues according to the studies: high levels were shown in almost hematopoietic cells and tissues, particularly in CD8+ and CD4+ T lymphocytes, B lymphocytes, Natural Killer (NK) cells and NK-T [19[19][20][21][22][23],20,21,22,23], whereas intermediate levels were shown in macrophages and dendritic cells [21,24,25,26][21][24][25][26]. Upregulated MHC-I expression is performed not only through the NLRC5 pathway itself, but also in a synergistic way with the help of additional cis-regulatory elements’ (ISRE and enhancer A) activation. Their expression is induced by IFNγ-caused STAT1 homodimer formation, which activates IRF1 and NLRC5 gene promoters through binding gamma-activated sequence (GAS) [27]. This was also confirmed by the involvement of activated IFN and STAT1 in the increase in NLRC5 expression during the activation of CTLs and T helper (Th) cells [21]. In vitro, the efficiency of TCR transgenic CD8+ T cells was activated and supported by NLRC5-deficient bone marrow-derived macrophages (BMDM) provided with SINFEKKL peptide, which was as similar as the efficacy of CD8+ T cells induced by wild-type BMDCs, while it was failed with the application of NLRC5-deficient cells provided with negative control [28]. After in vivo intravenous loading of Listeria monocytogenes, the IFN-producing CD8+ T cell was not aroused to an increase in the cell number in the spleen or the liver of NLRC5-deficient mice, consequently resulting in severe infection compared with the CD8+ T cell in wild mice [28,29][28][29]. In addition to CD8+ and CD4+ T lymphocytes, NLRC5 is implicated in γδT cell activation as well and its expression positively correlated with the induction of butyrophilin (BTN) family protein BTN3A1-3, whose gene contains SXY modules W/S, X1, X2 and Y boxes as cis-regulatory elements of the MHC-II gene promoter [30]. All these studies suggest that NLRC5 plays a role in the activation of MHC-I transcription, and the facilitation of CD8+ and CD4+ T lymphocyte development and immune function initiation (killing or helper).
In spite of numbers of studies proving NLRC5 expression in different tissues and its role in the MHC-I antigen presentation pathway [19,20,21,22[19][20][21][22][23],23], the regulatory mechanisms of the expression have not been well-elicited. These studies suggest that Chromatin NLRC 5 promoter accessibility is varied and modulated by different transcription factors in different cell types, where epigenetic regulation of chromatin remodeling may be a key pathway potentially adjusting NLRC 5 expression, such as in human cancers as well as cancer cell lines. NLRC 5 was involved in MHC-I expression attenuation through regulating promoter hypermethylation [18]. NLRC 5 expression, like other functional proteins, can also be regulated at the post-transcriptional and post-translational levels by long non-coding RNAs (lncRNAs) [31,32,33,34,35,36][31][32][33][34][35][36]. Interestingly, it was recently reported that MHC-I and APM expression was reduced by depressing NLRC5 in quiescent hair follicle stem cells and muscle stem cells in order to protect from immune surveillance and destruction [37]. Additionally, the reduction of MHC-I in cancer-originating cells of different types of cancer disrupted CTL-mediated damage, leading to the formation of variants that could escape immune surveillance and thereafter causing cancer recurrence [38,39,40,41,42,43][38][39][40][41][42][43]. Compared with immune evasion, these may suggest that NLRC5 and MHC-I reduction may facilitate the formation of a global dedifferentiation phenotype in cancer cells, which suggests the progression of cancer growth and the initiation of cancer cell communities.
As NLRC5 plays key role in the activation of MHC-I expression which is commonly lost in cancer immune evasion, NLRC5 could be the point that can switch on antitumor immunity. Several relevant in vitro and in vivo studies suggested that MHC-I expression in tumor cells could be more necessary than that expression in APCs to induce protective antitumor immunity [44,45][44][45]. Therefore, NLRC5 expression acquisition in tumor cells can enhance their MHC-I expression and antigen presentation and put forward antitumor immune response and CTL-mediated destruction [46,47,48][46][47][48]. However, in contradiction with the studies above focusing on the antitumor aspects of NLRC5, there are also studies implicating the role of NLRC5 in promoting tumor growth. According to their reports, NLRC5 was shown to participate in vitro in modulating the biological behaviors of HCC, ccRCC, glioma and ESCC cells, such as promoting cancer cell growth, motility and migration, and played a similar role in mice lacking the functionality of an adaptive immune system [33[33][36][49][50][51][52][53],36,49,50,51,52,53], while NLRC5 in mice with a competent immune system evidently suppressed the growth of melanoma and pancreatic ductal adenocarcinoma (PDAC) cells [54,55][54][55]. Considering that NLRC5 and MHC-I could be involved in progressive cancer growth and cancer cell population initiation, those contrary phenomena may result from that NLRC5-elicited antitumor immune response overcoming NLRC5-stimulated cancer growth in cancer cells originating from mice with normal immune function.
Studies on how to compensate for insufficient MHC-I expression and blocked antigen presentation in tumor cells are still progressing. IFN has been shown to potentially reverse the defect in MHC-I expression and accelerate antigen processing in cancer cells through the modification of proteasome constituents [45]. However, the limitation of potential use of IFN is there, where its systemic toxicity and development of cancer unresponsiveness to IFN are involved [56,57,58][56][57][58]. Epigenetic drugs may derepress NLRC5 showing the capacity to restore MHC-I expression, while their efficacy on solid tumors is limited and off-target effect is also a key challenge [59,60,61][59][60][61]. In general, genetic and epigenetic alteration-caused loss of NLRC5 paves the way for MHC-I deficiency, which is a common mechanism of cancer unresponsiveness to immunotherapy.
References
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274.
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10.
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80.
- Parham, P.; Lomen, C.E.; Lawlor, D.A.; Ways, J.P.; Holmes, N.; Coppin, H.L.; Salter, R.D.; Wan, A.M.; Ennis, P.D. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc. Natl. Acad. Sci. USA 1988, 85, 4005–4009.
- Bjorkman, P.J.; Parham, P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 1990, 59, 253–288.
- Shawar, S.M.; Vyas, J.M.; Rodgers, J.R.; Rich, R.R. Antigen presentation by major histocompatibility complex class I-B molecules. Annu. Rev. Immunol. 1994, 12, 839–880.
- D’Souza, M.P.; Adams, E.; Altman, J.D.; Birnbaum, M.E.; Boggiano, C.; Casorati, G.; Chien, Y.H.; Conley, A.; Eckle, S.B.G.; Fruh, K.; et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLoS Pathog. 2019, 15, e1007567.
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186.
- Koopman, L.A.; Corver, W.E.; van der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J. Exp. Med. 2000, 191, 961–976.
- Paschen, A.; Mendez, R.M.; Jimenez, P.; Sucker, A.; Ruiz-Cabello, F.; Song, M.; Garrido, F.; Schadendorf, D. Complete loss of HLA class I antigen expression on melanoma cells: A result of successive mutational events. Int. J. Cancer 2003, 103, 759–767.
- Bukur, J.; Jasinski, S.; Seliger, B. The role of classical and non-classical HLA class I antigens in human tumors. Semin. Cancer Biol. 2012, 22, 350–358.
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 279.
- Campoli, M.; Chang, C.C.; Ferrone, S. HLA class I antigen loss, tumor immune escape and immune selection. Vaccine 2002, 20 (Suppl. S4), A40–A45.
- Cabrera, T.; Lara, E.; Romero, J.M.; Maleno, I.; Real, L.M.; Ruiz-Cabello, F.; Valero, P.; Camacho, F.M.; Garrido, F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol. Immunother. CII 2007, 56, 709–717.
- Seliger, B.; Ruiz-Cabello, F.; Garrido, F. IFN inducibility of major histocompatibility antigens in tumors. Adv. Cancer Res. 2008, 101, 249–276.
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885.
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; van den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799.
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004.
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496.
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691.
- Staehli, F.; Ludigs, K.; Heinz, L.X.; Seguin-Estevez, Q.; Ferrero, I.; Braun, M.; Schroder, K.; Rebsamen, M.; Tardivel, A.; Mattmann, C.; et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J. Immunol. 2012, 188, 3820–3828.
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grotzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000.
- Neerincx, A.; Rodriguez, G.M.; Steimle, V.; Kufer, T.A. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J. Immunol. 2012, 188, 4940–4950.
- Robbins, G.R.; Truax, A.D.; Davis, B.K.; Zhang, L.; Brickey, W.J.; Ting, J.P. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J. Biol. Chem. 2012, 287, 24294–24303.
- Lupfer, C.R.; Stokes, K.L.; Kuriakose, T.; Kanneganti, T.D. Deficiency of the NOD-Like Receptor NLRC5 Results in Decreased CD8(+) T Cell Function and Impaired Viral Clearance. J. Virol. 2017, 91, e00377-17.
- Sun, T.; Ferrero, R.L.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. NLRC5 deficiency has a moderate impact on immunodominant CD8(+) T cell responses during rotavirus infection of adult mice. Immunol. Cell Biol. 2019, 97, 552–562.
- Kobayashi, K.S. NLRC5/CITA: A novel regulator of class I major histocompatibility complex genes. J. Immunodefic. Disord. 2012, 1, 1000e102.
- Biswas, A.; Meissner, T.B.; Kawai, T.; Kobayashi, K.S. Cutting edge: Impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J. Immunol. 2012, 189, 516–520.
- Yao, Y.; Wang, Y.; Chen, F.; Huang, Y.; Zhu, S.; Leng, Q.; Wang, H.; Shi, Y.; Qian, Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012, 22, 836–847.
- Dang, A.T.; Strietz, J.; Zenobi, A.; Khameneh, H.J.; Brandl, S.M.; Lozza, L.; Conradt, G.; Kaufmann, S.H.E.; Reith, W.; Kwee, I.; et al. NLRC5 promotes transcription of BTN3A1-3 genes and Vgamma9Vdelta2 T cell-mediated killing. iScience 2021, 24, 101900.
- Zhang, P.; Yu, C.; Yu, J.; Li, Z.; Lan, H.Y.; Zhou, Q. Arid2-IR promotes NF-kappaB-mediated renal inflammation by targeting NLRC5 transcription. Cell Mol. Life Sci. 2020, 78, 2387–2404.
- Zhou, Q.; Huang, X.R.; Yu, J.; Yu, X.; Lan, H.Y. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1034–1043.
- Zong, Z.; Song, Y.; Xue, Y.; Ruan, X.; Liu, X.; Yang, C.; Zheng, J.; Cao, S.; Li, Z.; Liu, Y. Knockdown of LncRNA SCAMP1 suppressed malignant biological behaviours of glioma cells via modulating miR-499a-5p/LMX1A/NLRC5 pathway. J. Cell Mol. Med. 2019, 23, 5048–5062.
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFkappaB signaling axis. Brain Behav. Immun. 2019, 80, 227–237.
- Li, J.; Yu, L.; Shen, Z.; Li, Y.; Chen, B.; Wei, W.; Chen, X.; Wang, Q.; Tong, F.; Lou, H.; et al. miR-34a and its novel target, NLRC5, are associated with HPV16 persistence. Infect. Genet. Evol. 2016, 44, 293–299.
- Zong, Y.; Zhang, Y.; Hou, D.; Xu, J.; Cui, F.; Qin, Y.; Sun, X. The lncRNA XIST promotes the progression of breast cancer by sponging miR-125b-5p to modulate NLRC5. Am. J. Transl. Res. 2020, 12, 3501–3511.
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.
- Schatton, T.; Schutte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708.
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 800–813.
- Maccalli, C.; Volonte, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655.
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360.
- Sultan, M.; Coyle, K.M.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis 2017, 38, 107–118.
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469.
- Rodriguez, G.M.; Bobbala, D.; Serrano, D.; Mayhue, M.; Champagne, A.; Saucier, C.; Steimle, V.; Kufer, T.A.; Menendez, A.; Ramanathan, S.; et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8(+) T lymphocytes. Oncoimmunology 2016, 5, e1151593.
- Seliger, B.; Wollscheid, U.; Momburg, F.; Blankenstein, T.; Huber, C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001, 61, 1095–1099.
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz5683.
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STINGand Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376.
- Kalbasi, A.; Tariveranmoshabad, M.; Hakimi, K.; Kremer, S.; Campbell, K.M.; Funes, J.M.; Vega-Crespo, A.; Parisi, G.; Champekar, A.; Nguyen, C.; et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl. Med. 2020, 12, eabb0152.
- Peng, Y.Y.; He, Y.H.; Chen, C.; Xu, T.; Li, L.; Ni, M.M.; Meng, X.M.; Huang, C.; Li, J. NLRC5 regulates cell proliferation, migration and invasion in hepatocellular carcinoma by targeting the Wnt/beta-catenin signaling pathway. Cancer Lett. 2016, 376, 10–21.
- Wang, Q.; Ding, H.; He, Y.; Li, X.; Cheng, Y.; Xu, Q.; Yang, Y.; Liao, G.; Meng, X.; Huang, C.; et al. NLRC5 mediates cell proliferation, migration, and invasion by regulating the Wnt/beta-catenin signalling pathway in clear cell renal cell carcinoma. Cancer Lett. 2019, 444, 9–19.
- He, Y.H.; Li, M.F.; Zhang, X.Y.; Meng, X.M.; Huang, C.; Li, J. NLRC5 promotes cell proliferation via regulating the AKT/VEGF-A signaling pathway in hepatocellular carcinoma. Toxicology 2016, 359–360, 47–57.
- Fan, Y.; Dong, Z.; Shi, Y.; Sun, S.; Wei, B.; Zhan, L. NLRC5 promotes cell migration and invasion by activating the PI3K/AKT signaling pathway in endometrial cancer. J. Int. Med. Res. 2020, 48, 300060520925352.
- Hu, X.; Wang, M.; Cao, L.; Cong, L.; Gao, Y.; Lu, J.; Feng, J.; Shen, B.; Liu, D. miR-4319 Suppresses the Growth of Esophageal Squamous Cell Carcinoma Via Targeting NLRC5. Curr. Mol. Pharm. 2020, 13, 144–149.
- Arnett, H.A.; Viney, J.L. Immune modulation by butyrophilins. Nat. Rev. Immunol. 2014, 14, 559–569.
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503.
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322.
- Xi, S.; Dyer, K.F.; Kimak, M.; Zhang, Q.; Gooding, W.E.; Chaillet, J.R.; Chai, R.L.; Ferrell, R.E.; Zamboni, B.; Hunt, J.; et al. Decreased STAT1 expression by promoter methylation in squamous cell carcinogenesis. J. Natl. Cancer Inst. 2006, 98, 181–189.
- Komyod, W.; Bohm, M.; Metze, D.; Heinrich, P.C.; Behrmann, I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol. Cancer Res. 2007, 5, 271–281.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. Ther. 2019, 4, 62.
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113.
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362.
More