Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Rita Xu and Version 2 by Rita Xu.
Almost 17% of Western patients affected by non-small cell lung cancer (NSCLC) have an activating epidermal growth factor receptor (EGFR) gene mutation. Del19 and L858R are the most-common ones; they are positive predictive factors for EGFR tyrosine kinase inhibitors (TKIs). Osimertinib, a third-generation TKI, is the standard first-line therapy for advanced NSCLC patients with common EGFR mutations.
- EGFR mutations
- non-small cell lung cancer
- tyrosine kinase inhibitors
1. Introduction
Non-small cell lung cancer (NSCLC) is the most-frequent cause of cancer-related deaths in the world [1]. Platinum-based chemotherapy was the only therapeutic option for advanced NSCLC patients for many years with a poor prognosis because of a median overall survival (OS) < 12 months [2]. However, the discovery of NSCLC oncogenic drivers led to the development of targeted drugs with an impressive survival benefit for select patients. In particular, the most-important oncogenic drivers are the epidermal growth factor receptor (EGFR) gene mutations [3]. Currently, various EGFR tyrosine kinase inhibitors (EGFR-TKIs) are standard treatment options for patients with activating EGFR gene mutations.
1.1. Epidermal Growth Factor Receptor Pathway in NSCLC
EGFR (ERBb1/HER1) belongs to the HER (ERBb) family with three other members: HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [4]. The binding of specific ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), to the EGFR extracellular domain led to receptor dimerisation with other HER family members [5]. Consequently, the autophosphorylation at the receptor key tyrosine residues takes place. In this way, various downstream signalling pathways are activated including the rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) pathway, the phospholipase C-protein kinase C (PLC-PKC) pathway, and the janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, which regulate cellular proliferation, survival, and apoptosis [6]. EGFR exons 18 to 24 encode the tyrosine kinase domain of the receptor. EGFR-activating gene mutations are located in exons 18, 19, 20, and 21 (the most-common ones in exons 19 and 21) and are responsible for the constitutive EGFR activation, which leads to cell proliferation [7]. These mutations are present in 10–15% of Caucasian NSCLC patients and 30–50% of Asian ones. They regard a more typically adenocarcinoma histotype, women, non-smokers, or Asian NSCLC patients [3].
The EGFR-activating gene mutations in exons 18, 19, 20, and 21 are classically divided into common (exon 19 deletion, exon 21 L858R point mutation), which correspond to 85–90% and generally confer sensitivity to EGFR-TKIs treatment, and uncommon (rare EGFR mutations and complex EGFR mutations), which account for 10–15% and present variable predictive values, from sensitivity to resistance [4][8]. Moreover, it is also possible to find other EGFR alterations consisting of the combination of EGFR mutations with other EGFR mutations or with one or more mutations of other genes (tumour suppressor gene or oncogene). In some cases, only a small percentage of tumour cells has the specific EGFR mutation. The variable sensitivities to EGFR-TKIs could be explained by the variable tertiary structure of the EGFR protein under the influence of the different EGFR mutations [8].
1.1.1. Common EGFR mutations
Several studies reported that Del19 (45–50%) appears more frequently than L858R (37–40%) in Asian, as well as in Caucasian populations [4].
The Del19 and L858R mutations lead to elevated receptor dimerisation and activity due to the destabilisation of the inactive conformation of the EGFR receptor [9].
While the L858R mutation corresponds to the substitution of leucine to arginine at codon 858 (c.2573T > G, p.L858R), Del19 presents more than 30 variants, and the most-common is delE746_A750 corresponding to 73% of cases, with a deletion of 9 to 24 nucleotides [9]. In 25% of cases, Del19 variants start at position E747. The remaining percentage is known as entitled non-LRE (2%) [5][9].
1.1.2. Uncommon EGFR Mutations
Approximatively 600 EGFR uncommon or rare mutations have been described, accounting for 10–15% of cases, with variable sensitivity to EGFR-TKI treatment [10] and a similar clinical presentation in comparison with common ones. Examples of rare EGFR mutations are some EGFR exon point mutations such as exon 18, G719X; exon 20, S768I; and exon 21, L861Q [4][11].
1.1.3. Other EGFR Alterations
Complex mutations have a prevalence of 5–15% of all EGFR mutations. They are defined by the combination of common and rare EGFR mutations or rare and rare EGFR mutations or common and common ones. The most-frequent EGFR mutations involved in complex mutations are G179X (90%), G709X (75%), and S768I (50%) [12][13][14][15]. The sensitivity to EGFR-TKIs depends on the specific combination of mutations. It is better when one of them is a sensitivity mutation, such as Del19 or L858R, and lower when the combination includes a resistance mutation. For example, E709A + G719C, G787R + L858R, H870R + L858R, and E884K + L858R are sensitive complex mutations, while T790M + L858R is a resistant one [12][13][14][15].
Co-mutations correspond to the combination of EGFR mutation(s) with one or more mutations of another gene (tumour suppressor gene or oncogene) [16]. Co-mutations account for similar prevalence across the common EGFR mutations. Their incidence seems to be correlated with prior treatment. These genetic alterations often are found in several genes such as TP53, RB1, CTNNB1 (β-catenin), NKX2-1, or PI3KCA [17][18][19]. Some co-mutations are correlated with a worse prognosis; for example, TP53 mutations, ATM alterations, PTEN-inactivating mutations, KRAS mutations, and IDH1 mutations are associated with lower clinical results following EGFR-TKI treatment [17].
Subclonal mutations have a low variant allele frequency (VAF), which may be due to the presence of the specific mutation only in a small percentage of tumour cells [20]. All types of EGFR mutations could be subject to these genetic alterations, in particular the resistant ones. For example, a retrospective analysis of the AURA study, the AURA3 trial, and the study performed by the French Cooperative Thoracic Intergroup showed that the T790 mutation was present only in a small proportion of patients with worse clinical outcomes under third-generation EGFR-TKIs [21][22][23][24].
1.2. Clinical Trials
EGFR-TKIs became the standard therapy for advanced EGFR-mutation-positive NSCLC patients after the evaluation of their safety and efficacy in several clinical trials performed in the last decade. Moreover, some clinical studies documented a better prognosis, in terms of progression-free survival (PFS) and OS, for Del19 compared to L858R-mutation NSCLC patients under treatment with EGFR-TKIs [25][26][27].
1.2.1. First-Generation EGFR-TKIs: Gefitinib, Erlotinib, and Icotinib
The NEJ002 study compared gefitinib versus carboplatin plus paclitaxel as a first-line therapy for advanced NSCLC patients with a common EGFR mutation [28][29].
The IPASS trial investigated gefitinib with carboplatin plus paclitaxel in the same population of the NEJ002 study [30][31].
WJTOG3405 is a phase 3 study in which common-EGFR-mutation NSCLC patients were randomised between gefitinib and cisplatin plus docetaxel [32][33].
The OPTIMAL trial evaluated erlotinib with respect to standard chemotherapy as a first-line therapy for common-EGFR-mutation NSCLC patients [34][35].
The ENSURE study analysed erlotinib in comparison with gemcitabine plus cisplatin in Asian patients affected by common-EGFR-mutation NSCLC [36].
In the EURTAC trial, erlotinib was compared to standard chemotherapy as a first-line treatment for European patients affected by common-EGFR-mutation NSCLC [37].
The CONVINCE study was designed to evaluate the efficacy and safety of icotinib as a first-line therapy compared to cisplatin/pemetrexed plus pemetrexed maintenance for common-EGFR-mutation NSCLC patients [38].
All these trials reported a significant improvement in terms of PFS, but no statistical difference was seen for OS, maybe because of the high percentage of crossover from standard therapy to the experimental one after disease progression.
1.2.2. Second-Generation EGFR-TKIs
The LUX-Lung 3 trial evaluated afatinib versus cisplatin plus gemcitabine or pemetrexed for EGFR-mutation NSCLC patients stratified according to mutation type (exon 19 deletion, L858R, or other) [39].
In the LUX-Lung 6 study, common-EGFR-mutation NSCLC patients were randomised between afatinib versus cisplatin plus gemcitabine or pemetrexed [40].
ARCHER 1050 investigated the safety and efficacy of dacomitinib with respect to gefitinib as a first-line treatment of advanced NCSLC patients with a common EGFR mutation [41].
All these trials reported a significant improvement in terms of PFS, but no statistical difference was seen for OS, maybe because of the high percentage of crossover from standard therapy to the experimental one after disease progression.
1.2.3. Third-Generation EGFR-TKI
AURA3 was designed to evaluate the safety and efficacy of osimertinib in comparison with cis/carboplatin plus pemetrexed for advanced NSCLC patients who experienced disease progression after first-line EGFR-TKI therapy and developed the EGFR T790 mutation. Indeed, osimertinib is the third-generation TKI selective for T790M resistance mutations. The authors reported a longer PFS (10.1 versus 4.4 months; HR 0.30; p < 0.001) and OS (26.8 versus 22.5 months; HR 0.87, p = 0.277) for the osimertinib group, although the latter had no significant difference. This is probably due to the high crossover rate from chemotherapy to osimertinib of patients with progressive disease. After crossover adjustment, there was an HR of 0.54 for OS. The ORR was significantly better with osimertinib (71%) than the control group (31%) (odds ratio for OR: 5.39; p < 0.001). Encephalic PFS was also significantly longer for patients treated with osimertinib (8.5 months vs. 4.2 months; HR 0.32) [42].
The FLAURA trial tested osimertinib versus standard EGFR-TKIs (gefitinib or erlotinib) in previously untreated patients with common-EGFR-mutation NSCLC. The results reported PFS and OS significantly longer for the osimertinib group (PFS: 18.9 months vs. 10.2 months; HR 0.46; p < 0.001. OS: 38.6 months vs. 31.8 months; HR 0.80 p = 0.046). The ORR was similar (80% with osimertinib and 76% with standard EGFR-TKIs; odds ratio: 1.27; p = 0.24) [26]. As a consequence of the good results reported in this trial, osimertinib has become the first-line treatment for advanced or metastatic EGFR-mutant-positive NSCLC, regardless of T790M status.
The ADAURA study analysed osimertinib for 3 years as an adjuvant therapy for NSCLC patients with stage IB-IIIA and common EGFR mutations who have or have not previously received adjuvant chemotherapy. The DFS rate was 73% and 38% at 4 years (HR 0.27; p < 0.001) for the overall population in the osimertinib and control group, respectively. As regards CNS disease, at 24 months, 98% and 85% of patients in the experimental and placebo group were alive and did not have central nervous system disease (HR 0.18). The OS results are still immature [43].
1.2.4. EGFR-TKIs Specific for Ins20
ZENITH20-2 is a multicentre, multicohort, open-label phase 2 trial that investigated poziotinib for previously treated advanced NSCLC patients with EGFR exon 20 insertions that demonstrated resistance to approved TKIs. This type of mutation is an oncogenic driver and accounts for 2–5% of NSCLCs. The ORR was 27.8%. The disease control rate (DCR) was 70.0%, and PFS was 5.5 months [44].
Mobocertinib was tested in a phase 1/2, dose-escalation and dose-expansion trial that enrolled pretreated patients with advanced NSCLC and EGFR exon 20 insertions. This drug is a TKI targeting EGFR exon 20 insertions in NSCLC. The ORR was 43%, and PFS was 7.3 months [45].
2. Mechanisms of Resistance
Although all generations of EGFR-TKIs have been proven to be very effective for NSCLC with common EGFR mutations, almost 5–25% of these patients do not experience a clinical benefit with these drugs due to intrinsic resistance [46][47]. On the other hand, the major part of patients treated with EGFR-TKIs became resistant to these therapies despite an initial response or stable disease. The various mechanisms of resistance to EGFR-TKIs could be explained by the high molecular heterogeneity of NSCLCs (Figure 1 and Figure 2) [48]. Therefore, deepening the knowledge about the EGFR-TKI resistance mechanisms is one of the most-important aims in order to improve the treatment strategy of these patients.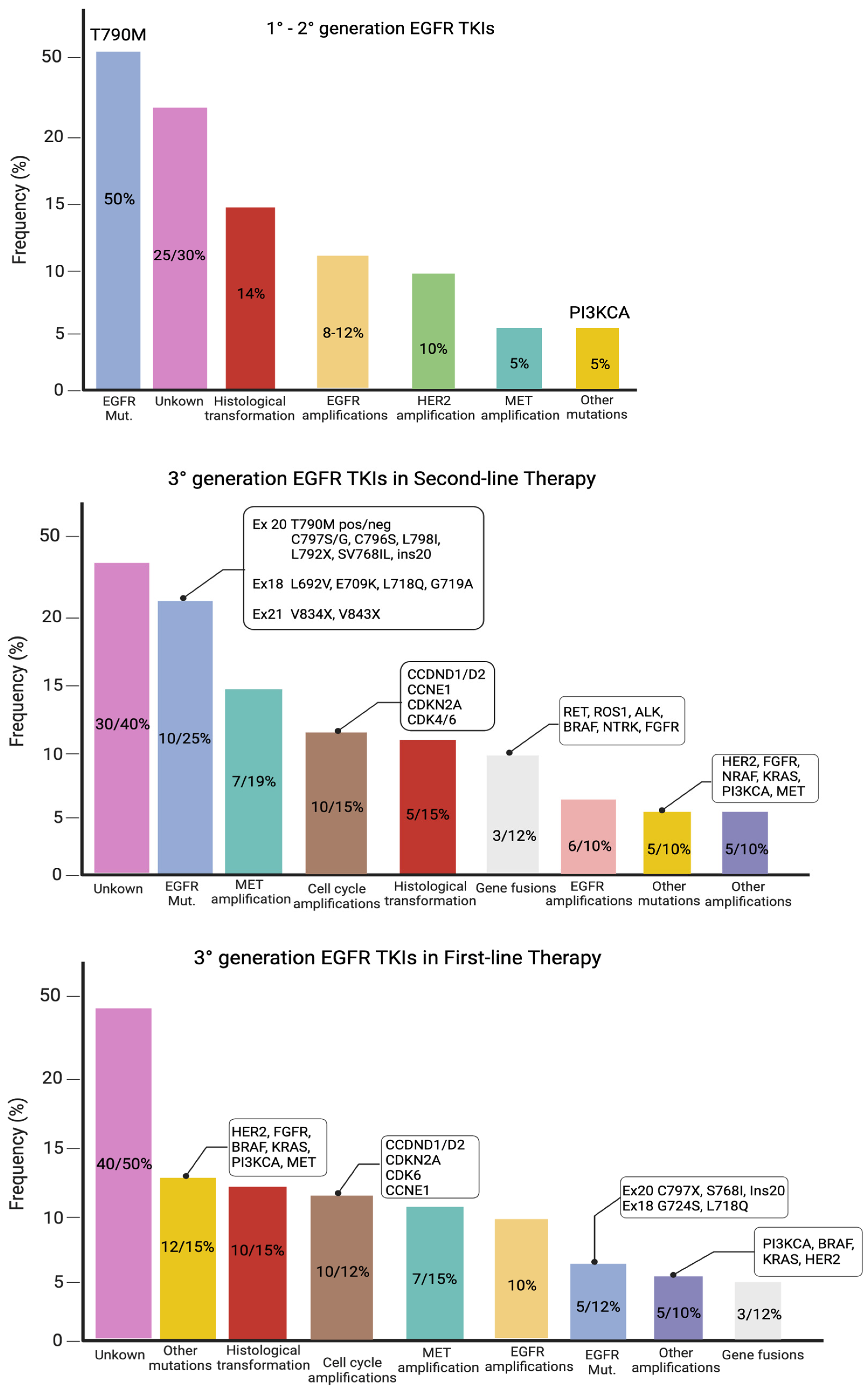
Figure 1. Mutations of resistance to EGFR-TKIs, according to the generation of TKIs and the line of therapy.
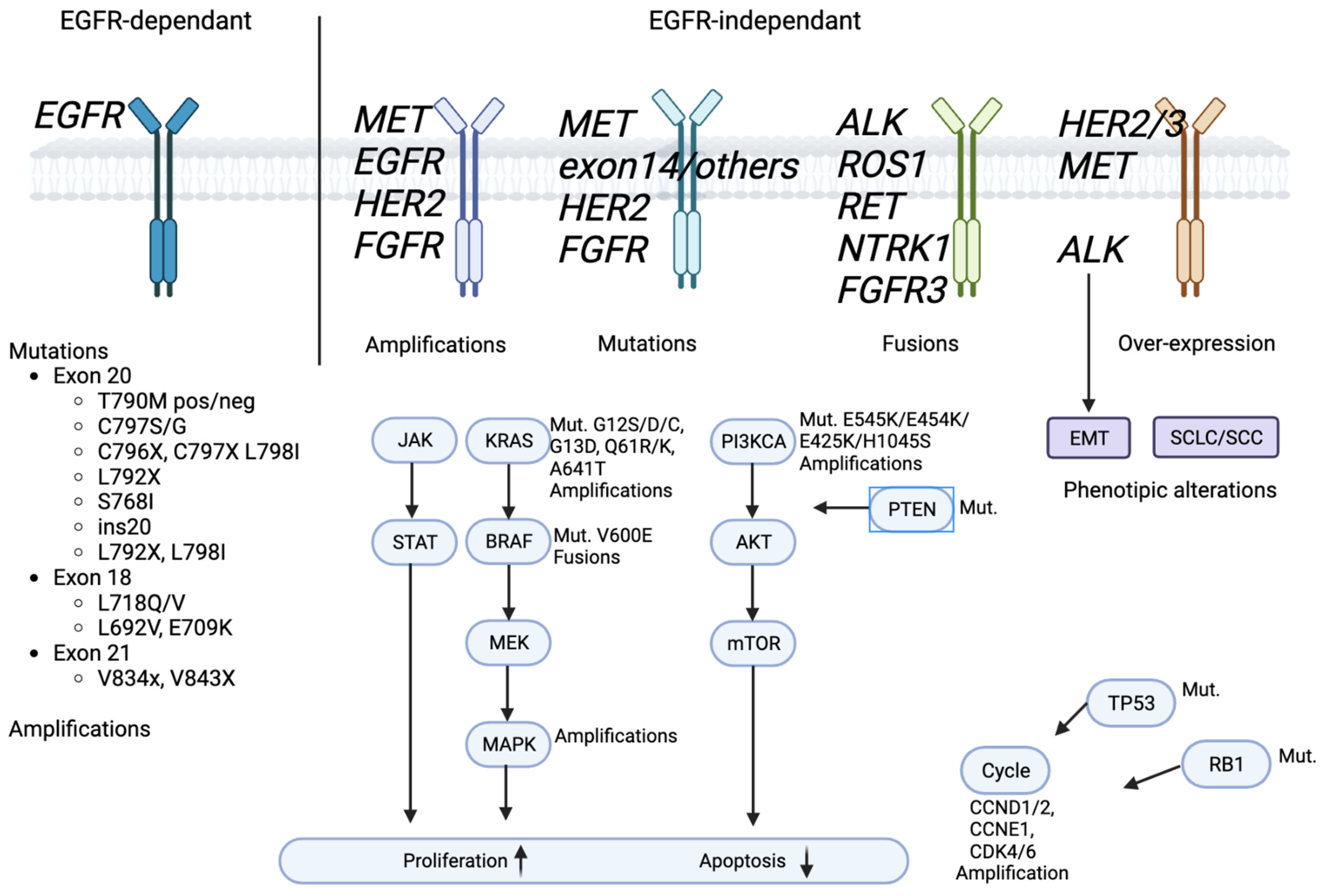
Figure 2. EGFR signaling pathway and EGFR-TKIs’ resistance mechanism.
2.1. Intrinsic Resistance
Patients with intrinsic resistance report an early tumour progression without prior tumour response; some of them respond for a very short period (<3 months) [49]. A possible cause regards the pharmacokinetics. In detail, treatment can fail due to the ineffective drug dose in the target area. This event can occur because of drug competition or the difficulty of first-/second-generation EGFR-TKIs to reach sanctuary localisations, such as the brain [50]. However, intrinsic resistance is often due to the lack of a target dependency (i.e., EGFR exon 20 mutations) or the genes alterations from other pathways (downstream or parallel pathways) [51][52]. Mechanisms of intrinsic resistance have been reported above all in patients with uncommon mutations and, more rarely, with common ones [51][52]. This type of resistance often depends on the presence of a drug-resistant EGFR mutation; the most-important ones are the exon 20 insertions (1–10% of all EGFR mutations) and the T790M EGFR mutation (approximately <1–65% of cases, based on the detection method employed) [53][54][55][56]. The lack of first-/second-generation EGFR-TKIs’ efficacy for the T790M mutation and exon 20 insertions is the reason for the development of third-generation EGFR-TKIs and EGFR-Ins20-specific inhibitors, respectively.2.1.1. Intrinsic Resistance to First-/Second-Generation EGFR-TKIs
Exon 20 insertions correspond to the addition of residues at the N-lobe of EGFR (M766 to C775), while the C-helix (A767 to C775) is their preferential location [57]. This area is fundamental to regulate ATP and EGFR-TKI binding with the consequent activation of the kinase domain through a conformation change [57]. Commonly, exon 20 insertion mutations led to a reduced sensitivity to EGFR-TKIs; however, in vitro studies described that some insertion mutations, such as the insertion EGFR-A763_Y764insFQEA, confer high sensitivity to EGFR-TKIs [58][59]. These data have been confirmed by clinical trials in which NSCLC patients with some types of insertion mutations experienced prolonged periods of disease control under EGFR-TKI treatment [58]. The presence of the EGFR T790M mutation at diagnosis is a rare event, which suggests, in some cases, a germinal EGFR mutation [55][56][60]. It is associated with the worst response to first- or second-generation EGFR-TKIs and poor clinical outcomes [55][56][61]. The EGFR T790M mutation regards exon 20 and consists of a substitution of the threonine at position 790 with a methionine in the ATP-binding pocket of the kinase domain. This change prevents the binding of EGFR-TKIs to the receptor due to a steric hindrance; on the other hand, it leads to an in increased affinity between ATP and EGFR. Therefore, the receptor affinity for ATP becomes greater than that for the drug with a severe reduction in EGFR-TKI activity [62]. Another EGFR mutation that is responsible for intrinsic resistance to EGFR-TKIs in vitro is the variant III (vIII) in-frame deletion of exons 2–7 in the extracellular domain [63]. This mutation is present in almost 5% of human lung squamous cell carcinoma and determines the unsuccessful binding of EGF and other growth factors to EGFRvIII [64][65]. The reason behind the constitutive activation of EGFRvIII and the EGFR-TKI resistance is probably the structural changes of the EGFR protein affecting the ATP pocket and the intracellular domain conformation [63]. Some genetic alterations could occur in NSCLC patients with common EGFR mutations with the consequent reduction of sensitivity to EGFR-TKI therapy. In this regard, BIM is a proapoptotic member of the Bcl-2 family that plays a critical role in apoptosis mediated by EGFR-TKIs [66]. NSCLC patients with deletion polymorphisms or low-to-intermediate levels of BIM mRNA have poor clinical efficacy under EGFR-TKIs [66][67]. Furthermore, low levels of NF1 and the overexpression of RhoB are correlated with poor clinical efficacy [68][69]. Moreover, the plasma detection of TP53 gene co-mutations within two months of EGFR-TKI treatments is related to the worst PFS and OS [70]. CRIPTO1 is a member of the EGF-CFC family; it is a cell membrane protein linked to glycosylphosphatidylinositol. High basal levels of CRIPTO1 lead to a reduced EGFR-TKI sensitivity through the activation of ZEB1 and SRC. In this way, ZEB1 promotes epithelial-to-mesenchymal transition (EMT), while SCR stimulates AKT and MEK signalling [71]. The major part of oncogenic driver mutations in NSCLC is mutually exclusive, although some of them are present simultaneously, such as PI3KCA or TP53 mutations, with some other oncogenic driver mutations [72][73]. If, on the one hand, co-mutations of the PI3KCA and EGFR genes have no clinical impact [72], on the other hand, the co-mutation of EGFR Del19 and non-disruptive TP53 exon 8 is correlated with intrinsic resistance to first-generation EGFR-TKIs [73]. In some cases, pretreatment AXL and CDCP1 RNA overexpression coexist with EGFR mutations and correlate with poor response to first-generation EGFR-TKIs [74] and, likewise, co-alterations in some cell cycle genes or genes of the PI3K, MAPK, and Wnt/β-catenin pathways [16][49].2.1.2. Intrinsic Resistance to Third-Generation EGFR-TKIs
Although most studies regard resistance to osimertinib during second-line therapy, some literature data report intrinsic resistance when it is administered as a first-line treatment [75]. In this regard, the transformation of NSCLC to SCLC has been considered a possible mechanism of intrinsic resistance [76]. The HER2 and MET genes’ amplification was associated in in vitro studies with reduced sensitivity to third-generation EGFR-TKIs such as osimertinib and rolecitinib [77][78]. The combination of the KRAS G12D mutation and PTEN loss was also detected in NSCLC patients with intrinsic resistance to osimertinib [49]. The worst response to third-generation EGFR-TKIs was reported also in NSCLC patients with the EGFR mutation and CDCP1 or AXL RNA overexpression at baseline [74].2.2. Acquired Resistance
All the patients treated with EGFR-TKIs experience a progression of disease (PD) after a variable period of treatment. Patients usually develop acquired resistance after 9–12 months of treatment with first-/second-generation EGFR-TKIs, almost 10 months with second-line third-generation EGFR-TKIs [42], and about 19 months with first-line third-generation EGFR-TKIs [27]. Clinical criteria of acquired resistance to EGFR-TKIs in NSCLC patients have been proposed by Jackman et al., although further clinical validation is needed. The criteria by Gandara et al. are based on the type of PD: central nervous system (CNS), systemic, and oligo-progression. In the consideration of the undefined management of NSCLC patients who progressed to EGFR-TKIs because of acquired resistance, this classification could help clinicians establish the best treatment strategy based on the PD patterns [77]. For example, the same treatment with EGFR-TKI could be continued in patients with slow-PD and without clinical deterioration. A similar strategy could be applied for those patients with CNS PD or oligo-PD in association with local treatment to the site of progression (e.g., radiotherapy or surgery) [79]. The comprehension of the mechanisms leading to the acquired resistance is complex due to different aspects such as: (1) the type of EGFR-TKI; (2) the line of treatment with a specific EGFR-TKI; (3) the tumour biology, in particular histology, intrinsic mutability, microenvironment, and the type of initial EGFR mutation. In the literature, there are several studies regarding the development of acquired resistance to the first-line treatment with first- or second-generation EGFR-TKIs or second-line treatment with third-generation EGFR-TKIs, usually due to the occurrence of the T790M mutation. In contrast, few data have been published about the acquired resistance to first-line osimertinib treatment. However, given the increasingly larger number of patients who will be treated with this drug, it is crucial to deepen the knowledge about the biological mechanisms of EGFR-TKI resistance. Below, wresearchers describe the acquired resistance mechanisms to EGFR-TKIs known today. In detail, they can be classified into EGFR-dependent due to the insurgence of new EGFR mutations and EGFR-independent mechanisms due to the activation of alternative pathways.2.2.1. EGFR-Dependent Mechanisms: Secondary EGFR Mutations
Acquired resistance based on EGFR-dependent mechanisms is due to the insurgence of secondary and tertiary mutations and/or amplifications of the EGFR gene with the consequent alteration of the receptor aminoacidic structure. Therefore, this leads to a conformational change that can regard the kinase or the ATP-binding pocket of the mutant EGFR, limiting drug accessibility or increasing the ATP affinity. The incidence of EGFR-dependent acquired resistance is variable based on the type of EGFR-TKI administered and the line of treatment. To be specific, approximately 50% of patients develop this type of resistance under first-/second-generation EGFR-TKIs, 20% of them if they are treated with third-generation EGFR-TKI as a second-line therapy, and 10–15% with first-line third-generation EGFR-TKIs [80].EGFR T790 Mutation
This is the most-frequent (49–63%) secondary mutation resulting in the insurgence of acquired resistance under treatment with first-/second-generation EGFR-TKIs [81]. Therefore, third-generation inhibitors were specifically designed to target the EGFR T790 mutation. The EGFR T790M mutation regards exon 20 and consists of a substitution of the threonine at position 790 with a methionine in the ATP-binding pocket of the kinase domain. This change prevents the binding of EGFR-TKIs to the receptor due to a steric hindrance; on the other hand, it leads to an increased affinity between ATP and EGFR. Therefore, the receptor affinity for ATP becomes greater than that for the drug with a severe reduction in EGFR-TKI activity [82]. Some studies have hypothesised that this type of resistance might depend on the selection of pre-existing drug-resistant EGFR-T790M-positive clones during treatment with first-/second-generation EGFR-TKIs or on de novo acquisition of the EGFR-T790M mutation by initially drug-tolerant cells, negative for the EGFR T790M mutation [83]. Experimental data on gefitinib-resistant PC9 cells showed that the early EGFR T790M mutant clones derived from pre-existing EGFR T790M mutated cells were selected for gefitinib treatment. The other theory includes the late de novo occurrence of this type of mutation in drug-tolerant cells due to the prolonged exposure to a first-/second-generation EGFR inhibitor [53]. In an in vitro study, Hata et al. documented the restorations of late-emerging T790M cells’ sensitivity to third-generation EGFR-TKIs thanks to the treatment of tumour cells with navitoclax, an inhibitor of the antiapoptotic factors BCL-2 and BCL-xL [84]. Approximately 43% of NSCLC patients lose the EGFR T790M mutation with the PD [30][53][57]. This event suggests the existence of subclones with the EGFR T790M mutation [77]. Usually, the loss of T790M is associated with the presence of exon 19 deletion (83%) and, only rarely, with the L858R mutation (14%) [57]. Moreover, from a clinical point of view, this event at the time of progression is correlated with the worst clinical outcomes [23][85][86][87]. From a molecular point of view, it is associated with the loss of EGFR dependence and dependence on non-EGFR mechanisms [85].Tertiary EGFR Mutations: Resistance to Second-Line Third-Generation EGFR-TKI
The AURA3 trial was the first study that showed the insurgence of acquired resistance to second-line osimertinib by means of the employment of cell-free DNA (cfDNA) genomic profiles [85][88]. To be specific, the results demonstrated that about 50% of the NSCLC patients maintained the EGFR T790M mutation, including those that experienced the insurgence of the tertiary EGFR mutation. The authors reported that acquired tertiary EGFR mutations occurred in 21% of cases, and the most-common one (15%) was the EGFR exon 20 C797S mutation [42][89][90]. In contrast, the FLAURA study, in which osimertinib was administered as a first-line therapy, reported a C797S mutation frequency of 7%. The C797S mutation corresponds to a substitution in the ATP-binding site of a cysteine with a serine at codon 797, resulting in the inability of osimertinib to covalently bond with the mutant EGFR [91]. Moreover, some studies showed that this mutation also prevents the binding of other irreversible third-generation EGFR-TKIs such as olmutinib, rociletinib, and narzatinib to the EGFR active site [92][93]. Interestingly, the allelic context of the C797S mutation can predict the response to subsequent EGFR-TKI therapies. In detail, when NSCLC patients have the T790M and C797S mutations on the same allele (cis-mutations), they experience resistance to all available generations of EGFR-TKIs as a single agent or combined with other drugs [93][94]. On the other hand, when patients have these mutations on different alleles (trans-mutations), they experience sensitivity to first- and second-generation EGFR-TKIs. However, mutations in trans are rare, regarding less than 30% of cases [94][95]. Rare point EGFR mutations in exon 20 have been also identified in the C796 residue such as the G796R (0.56% of patients with lung adenocarcinoma treated with osimertinib), G796S, and G796D mutations, which are adjacent to C797 in exon 20 and can sterically impair the binding of osimertinib to EGFR [92][96][97][98]. L792 exon 20 mutations, including L792H, L792Y, and L792F, consist of the addition of a benzene or imidazole ring to the side chain of L792, resulting in the binding disruption of osimertinib to the EGFR kinase domain [97][99]. This mutation usually is located in cis with T790M, but less frequently, it can also occur in trans with EGFR C796/C797X mutations [94]. The L718Q, L718V, and L798I mutations in exon 18 affect the ATP-binding site of the EGFR kinase domain. Therefore, they determine steric restriction, preventing the binding of osimertinib [86][94][99]. These mutations are responsible for osimertinib resistance independent of the C797 mutation; in fact, they are not co-existent. L718Q/V are associated with sensitivity to first- and second-generation EGFR-TKIs, above all when T790M has been lost [100]. Osimertinib resistance is also caused by the G719A mutation located close to the L718 residue [94]. G724S is a very rare EGFR mutation located in exon 20, usually associated with EGFR exon 19 deletion. This mutation regards the P-loop of the kinase domain interfering with the binding of osimertinib [101][102][103]. However, this altered structure does not confer resistance to second-generation EGFR inhibitors [51]. SV768IL (S768I + V769L) is another rare (3%) mutation of EGFR exon 20 that has been identified in second-line therapy with osimertinib [90]. Rarely, tertiary EGFR mutations such as G724S, L718Q, V834L, and L718V can occur in patients that lost the T790M mutation [86].Secondary EGFR Mutations: Resistance to First-Line Third-Generation EGFR-TKI
FLAURA was the first study that evaluated resistance to osimertinib as a first-line treatment [27][104]. Other literature data derive from some case reports or small case series [85][105]. This phase 3 trial analysed cfDNA samples through NGS, but no emergent T790M mutation has been detected. This discovery is in line with the well-known activity of osimertinib towards EGFR-sensitising and T790M mutations [104]. In this study, EGFR mutation/amplification was rare (9%), as well as C797S mutation frequency (7%), although it is the most-common mechanism after MET amplification (15%) [90]. S768I or combined EGFR mutation, such as Del19 + G724S (exon18), L718Q + EGFR ex20ins (exon 18 + 20), C797X, or S768I (exon 20), or L718Q + C797S, L718Q + L797S (exon 18 + 20) are very rare secondary mutations, each corresponding to about 1% of cases [11][12][104][106]. Interestingly, this restudyearch gave evidence that the mechanisms of resistance to first-line osimertinib depend on EGFR only for a small proportion of cases, and no EGFR T790M mutation was observed. Alternatively, EGFR-dependent mechanisms of acquired resistance are typical for those patients who receive osimertinib as a second-line therapy. Therefore, the T790M mutation will be less common due to the more-frequent use of osimertinib as the first-line therapy.Rare EGFR Mutations
Although the underlying mechanisms are not well-defined yet, literature data described other rare EGFR point mutations that are responsible for acquired resistance to first-/second-generation EGFR-TKIs and regard less than 10% of NSCLC patients. They include D761Y and L747S (exon 19) or T854A (exon 21), Asp761Tyr, 39 Thr854Ala, and 40 Leu747Ser [60][99][107]. Other rare molecular alterations, such as the β-catenin mutation, have been detected in association with the EGFR T790M mutation [81].References
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24.
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579.
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54.
- Leduc, C.; Merlio, J.P.; Besse, B.; Blons, H.; Debieuvre, D.; Bringuier, P.P.; Monnet, I.; Rouquette, I.; Fraboulet-Moreau, S.; Lemoine, A.; et al. Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: Results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann. Oncol. 2017, 28, 2715–2724.
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220.
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500.
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139.
- Bílek, O.; Holánek, M.; Berkovcová, J.; Horký, O.; Kazda, T.; Čoupková, H.; Špelda, S.; Kristková, L.; Zvaríková, M.; Podhorec, J.; et al. Uncommon EGFR Mutations in Non-Small Cell Lung Cancer and Their Impact on the Treatment. Klin. Onkol. Cas. Ceske A Slov. Onkol. Spol. 2019, 32, 6–12.
- Li, W.Q.; Cui, J.W. Non-small cell lung cancer patients with ex19del or exon 21 L858R mutation: Distinct mechanisms, different efficacies to treatments. J. Cancer Res. Clin. Oncol. 2020, 146, 2329–2338.
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431.
- Beau-Faller, M.; Prim, N.; Ruppert, A.M.; Nanni-Metéllus, I.; Lacave, R.; Lacroix, L.; Escande, F.; Lizard, S.; Pretet, J.L.; Rouquette, I.; et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: A multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 2014, 25, 126–131.
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179.
- Kobayashi, S.; Canepa, H.M.; Bailey, A.S.; Nakayama, S.; Yamaguchi, N.; Goldstein, M.A.; Huberman, M.S.; Costa, D.B. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J. Thorac. Oncol. 2013, 8, 45–51.
- Lin, Y.T.; Tsai, T.H.; Wu, S.G.; Liu, Y.N.; Yu, C.J.; Shih, J.Y. Complex EGFR mutations with secondary T790M mutation confer shorter osimertinib progression-free survival and overall survival in advanced non-small cell lung cancer. Lung Cancer 2020, 145, 1–9.
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16.
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704.
- Blons, H.; Oudart, J.B.; Merlio, J.P.; Debieuvre, D.; de Fraipont, F.; Audigier-Valette, C.; Escande, F.; Hominal, S.; Bringuier, P.P.; Fraboulet-Moreau, S.; et al. PTEN, ATM, IDH1 mutations and MAPK pathway activation as modulators of PFS and OS in patients treated by first line EGFR TKI, an ancillary study of the French Cooperative Thoracic Intergroup (IFCT) Biomarkers France project. Lung Cancer 2021, 151, 69–75.
- Canale, M.; Petracci, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047.
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509.
- Kohsaka, S.; Petronczki, M.; Solca, F.; Maemondo, M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Future Oncol. 2019, 15, 637–652.
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment with Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382.
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical impact of subclonal EGFR T790M mutations in advanced-stage EGFR-mutant non-small-cell lung cancers. Nat. Commun. 2021, 12, 1780.
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534.
- Beau-Faller, M.; Pencreach, E.; Leduc, C.; Blons, H.; Merlio, J.-P.; Bringuier, P.-P.; de Fraipont, F.; Escande, F.; Lemoine, A.; Ouafik, L.H.; et al. Independent prognostic value of ultra-sensitive quantification of tumor pre-treatment T790M subclones in EGFR mutated non-small cell lung cancer (NSCLC) treated by first/second generation TKI, depends on variant allele frequency (VAF): Results of the French cooperative thoracic intergroup (IFCT) biomarkers France project. Lung Cancer 2020, 140, 19–26.
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186.
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50.
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125.
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59.
- Miyauchi, E.; Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Saijo, Y.; Yoshizawa, H.; et al. Efficacy of chemotherapy after first-line gefitinib therapy in EGFR mutation-positive advanced non-small cell lung cancer-data from a randomized Phase III study comparing gefitinib with carboplatin plus paclitaxel (NEJ002). Jpn. J. Clin. Oncol. 2015, 45, 670–676.
- Fukuoka, M.; Wu, Y.L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.S.; Sriuranpong, V.; Chao, T.Y.; Nakagawa, K.; Chu, D.T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874.
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957.
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128.
- Yoshioka, H.; Shimokawa, M.; Seto, T.; Morita, S.; Yatabe, Y.; Okamoto, I.; Tsurutani, J.; Satouchi, M.; Hirashima, T.; Atagi, S.; et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1978–1984.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883.
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246.
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE): A phase 3, open-label, randomized study. Ann. Oncol. 2017, 28, 2443–2450.
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151.
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222.
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466.
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640.
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723.
- Le, X.; Cornelissen, R.; Garassino, M.; Clarke, J.M.; Tchekmedyian, N.; Goldman, J.W.; Leu, S.Y.; Bhat, G.; Lebel, F.; Heymach, J.V.; et al. Poziotinib in Non-Small-Cell Lung Cancer Harboring HER2 Exon 20 Insertion Mutations after Prior Therapies: ZENITH20-2 Trial. J. Clin. Oncol. 2022, 40, 710–718.
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients with EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761.
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623.
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206.
- Dong, R.F.; Zhu, M.L.; Liu, M.M.; Xu, Y.T.; Yuan, L.L.; Bian, J.; Xia, Y.Z.; Kong, L.Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583.
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923.
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247.
- Jin, Y.; Shi, X.; Zhao, J.; He, Q.; Chen, M.; Yan, J.; Ou, Q.; Wu, X.; Shao, Y.W.; Yu, X. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer 2018, 124, 110–116.
- Morgillo, F.; Bareschino, M.A.; Bianco, R.; Tortora, G.; Ciardiello, F. Primary and acquired resistance to anti-EGFR targeted drugs in cancer therapy. Differ. Res. Biol. Divers. 2007, 75, 788–799.
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060.
- Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926.
- Attili, I.; Karachaliou, N.; Conte, P.; Bonanno, L.; Rosell, R. Therapeutic approaches for T790M mutation positive non-small-cell lung cancer. Expert Rev. Anticancer. Ther. 2018, 18, 1021–1030.
- Chen, L.Y.; Molina-Vila, M.A.; Ruan, S.Y.; Su, K.Y.; Liao, W.Y.; Yu, K.L.; Ho, C.C.; Shih, J.Y.; Yu, C.J.; Yang, J.C.; et al. Coexistence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: A systematic review and meta-analysis. Lung Cancer 2016, 94, 46–53.
- Eck, M.J.; Yun, C.-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 559–566.
- De Pas, T.; Toffalorio, F.; Manzotti, M.; Fumagalli, C.; Spitaleri, G.; Catania, C.; Delmonte, A.; Giovannini, M.; Spaggiari, L.; de Braud, F.; et al. Activity of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Patients with Non-small Cell Lung Cancer Harboring Rare Epidermal Growth Factor Receptor Mutations. J. Thorac. Oncol. 2011, 6, 1895–1901.
- Costa, D.B. More Than Just an Oncogene Translocation and a Kinase Inhibitor: Kevin’s Story. J. Clin. Oncol. 2011, 30, 110–112.
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180.
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Giménez-Capitán, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The Impact of EGFR T790M Mutations and BIM mRNA Expression on Outcome in Patients with EGFR-Mutant NSCLC Treated with Erlotinib or Chemotherapy in the Randomized Phase III EURTAC Trial. Clin. Cancer Res. 2014, 20, 2001–2010.
- Soejima, K.; Yasuda, H.; Hirano, T. Osimertinib for EGFR T790M mutation-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 31–38.
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822.
- Okamoto, I.; Kenyon, L.C.; Emlet, D.R.; Mori, T.; Sasaki, J.; Hirosako, S.; Ichikawa, Y.; Kishi, H.; Godwin, A.K.; Yoshioka, M.; et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003, 94, 50–56.
- Sasaki, H.; Kawano, O.; Endo, K.; Yukiue, H.; Yano, M.; Fujii, Y. EGFRvIII mutation in lung cancer correlates with increased EGFR copy number. Oncol. Rep. 2007, 17, 319–323.
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM Expression in Treatment-Naïve Cancers Predicts Responsiveness to Kinase Inhibitors. Cancer Discov. 2011, 1, 352–365.
- Murakami, H.; Nokihara, H.; Shimizu, T.; Seto, T.; Keating, A.; Krivoshik, A.; Uegaki, K.; Morita, S.; Nakagawa, K.; Fukuoka, M. 9LBA Antitumor activity of ASP8273, an irreversible mutant selective EGFR-TKI, in NSCLC patients with tumors harboring EGFR activating mutations and T790M resistance mutation. Eur. J. Cancer 2014, 50, 198.
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619.
- Calvayrac, O.; Mazières, J.; Figarol, S.; Marty-Detraves, C.; Raymond-Letron, I.; Bousquet, E.; Farella, M.; Clermont-Taranchon, E.; Milia, J.; Rouquette, I.; et al. The RAS-related GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO Mol. Med. 2017, 9, 238–250.
- Tsui, D.W.Y.; Murtaza, M.; Wong, A.S.C.; Rueda, O.M.; Smith, C.G.; Chandrananda, D.; Soo, R.A.; Lim, H.L.; Goh, B.C.; Caldas, C.; et al. Dynamics of multiple resistance mechanisms in plasma DNA during EGFR-targeted therapies in non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e7945.
- Park, K.-S.; Raffeld, M.; Moon, Y.W.; Xi, L.; Bianco, C.; Pham, T.; Lee, L.C.; Mitsudomi, T.; Yatabe, Y.; Okamoto, I.; et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J. Clin. Investig. 2014, 124, 3003–3015.
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719.
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202.
- Karachaliou, N.; Chaib, I.; Cardona, A.F.; Berenguer, J.; Bracht, J.W.P.; Yang, J.; Cai, X.; Wang, Z.; Hu, C.; Drozdowskyj, A.; et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-smallcell Lung Cancer Associated with Poor Prognosis. EBioMedicine 2018, 29, 112–127.
- Michels, S.; Heydt, C.; van Veggel, B.; Deschler-Baier, B.; Pardo, N.; Monkhorst, K.; Rüsseler, V.; Stratmann, J.; Griesinger, F.; Steinhauser, S.; et al. Genomic Profiling Identifies Outcome-Relevant Mechanisms of Innate and Acquired Resistance to Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy in Lung Cancer. JCO Precis. Oncol. 2019, 3, PO.18.00210.
- Minari, R.; Bordi, P.; Del Re, M.; Facchinetti, F.; Mazzoni, F.; Barbieri, F.; Camerini, A.; Comin, C.E.; Gnetti, L.; Azzoni, C.; et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer 2018, 115, 21–27.
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360.
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, I.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847.
- Gandara, D.R.; Li, T.; Lara, P.N.; Kelly, K.; Riess, J.W.; Redman, M.W.; Mack, P.C. Acquired Resistance to Targeted Therapies Against Oncogene-Driven Non–Small-Cell Lung Cancer: Approach to Subtyping Progressive Disease and Clinical Implications. Clin. Lung Cancer 2014, 15, 1–6.
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391.
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26.
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075.
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Lung Cancer: Distinct Natural History of Patients with Tumors Harboring the T790M Mutation. Clin. Cancer Res. 2011, 17, 1616–1622.
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-e.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269.
- Chmielecki, J.; Mok, T.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Shepherd, F.A.; et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat. Commun. 2023, 14, 1071.
- Fang, W.; Gan, J.; Huang, Y.; Zhou, H.; Zhang, L. Acquired EGFR L718V Mutation and Loss of T790M-Mediated Resistance to Osimertinib in a Patient with NSCLC Who Responded to Afatinib. J. Thorac. Oncol. 2019, 14, e274–e275.
- Lin, C.C.; Shih, J.Y.; Yu, C.J.; Ho, C.C.; Liao, W.Y.; Lee, J.H.; Tsai, T.H.; Su, K.Y.; Hsieh, M.S.; Chang, Y.L.; et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: A genomic study. Lancet Respir. Med. 2018, 6, 107–116.
- Papadimitrakopoulou, V.A.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; Delmonte, A.; Hsia, T.C.; Laskin, J.; Kim, S.W.; He, Y.; Tsai, C.M.; et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non-small cell lung cancer. Cancer 2020, 126, 373–380.
- Lu, X.; Yu, L.; Zhang, Z.; Ren, X.; Smaill, J.B.; Ding, K. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018, 38, 1550–1581.
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663.
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074.
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107.
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933.
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732.
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727.
- Li, Y.; Mao, T.; Wang, J.; Zheng, H.; Hu, Z.; Cao, P.; Yang, S.; Zhu, L.; Guo, S.; Zhao, X.; et al. Toward the next generation EGFR inhibitors: An overview of osimertinib resistance mediated by EGFR mutations in non-small cell lung cancer. Cell Commun. Signal. 2023, 21, 71.
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231.
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679.
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10.
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923.
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly emergent acquired EGFR exon 18 G724S mutation after resistance of a T790M specific EGFR inhibitor osimertinib in non-small-cell lung cancer: A case report. OncoTargets Ther. 2019, 12, 51–56.
- Oztan, A.; Fischer, S.; Schrock, A.B.; Erlich, R.L.; Lovly, C.M.; Stephens, P.J.; Ross, J.S.; Miller, V.; Ali, S.M.; Ou, S.I.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111, 84–87.
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin. Cancer Res. 2019, 25, 3341–3351.
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652.
- Ríos-Hoyo, A.; Moliner, L.; Arriola, E. Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge. Cancers 2022, 14, 1931.
- Kohsaka, S.; Nagano, M.; Ueno, T.; Suehara, Y.; Hayashi, T.; Shimada, N.; Takahashi, K.; Suzuki, K.; Takamochi, K.; Takahashi, F.; et al. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci. Transl. Med. 2017, 9, eaan6566.
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59.
More