Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Carmelo Laface and Version 3 by Rita Xu.
Almost 17% of Western patients affected by non-small cell lung cancer (NSCLC) have an activating epidermal growth factor receptor (EGFR) gene mutation. Del19 and L858R are the most-common ones; they are positive predictive factors for EGFR tyrosine kinase inhibitors (TKIs). Osimertinib, a third-generation TKI, is the standard first-line therapy for advanced NSCLC patients with common EGFR mutations.
- EGFR mutations
- non-small cell lung cancer
- tyrosine kinase inhibitors
1. Introduction
Non-small cell lung cancer (NSCLC) is the most-frequent cause of cancer-related deaths in the world [1]. Platinum-based chemotherapy was the only therapeutic option for advanced NSCLC patients for many years with a poor prognosis because of a median overall survival (OS) < 12 months [2]. However, the discovery of NSCLC oncogenic drivers led to the development of targeted drugs with an impressive survival benefit for select patients. In particular, the most-important oncogenic drivers are the epidermal growth factor receptor (EGFR) gene mutations [3]. Currently, various EGFR tyrosine kinase inhibitors (EGFR-TKIs) are standard treatment options for patients with activating EGFR gene mutations.
1.1. Epidermal Growth Factor Receptor Pathway in NSCLC
EGFR (ERBb1/HER1) belongs to the HER (ERBb) family with three other members: HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [4]. The binding of specific ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), to the EGFR extracellular domain led to receptor dimerisation with other HER family members [5]. Consequently, the autophosphorylation at the receptor key tyrosine residues takes place. In this way, various downstream signalling pathways are activated including the rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) pathway, the phospholipase C-protein kinase C (PLC-PKC) pathway, and the janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, which regulate cellular proliferation, survival, and apoptosis [6]. EGFR exons 18 to 24 encode the tyrosine kinase domain of the receptor. EGFR-activating gene mutations are located in exons 18, 19, 20, and 21 (the most-common ones in exons 19 and 21) and are responsible for the constitutive EGFR activation, which leads to cell proliferation [7]. These mutations are present in 10–15% of Caucasian NSCLC patients and 30–50% of Asian ones. They regard a more typically adenocarcinoma histotype, women, non-smokers, or Asian NSCLC patients [3].
The EGFR-activating gene mutations in exons 18, 19, 20, and 21 are classically divided into common (exon 19 deletion, exon 21 L858R point mutation), which correspond to 85–90% and generally confer sensitivity to EGFR-TKIs treatment, and uncommon (rare EGFR mutations and complex EGFR mutations), which account for 10–15% and present variable predictive values, from sensitivity to resistance [4][8][4,8]. Moreover, it is also possible to find other EGFR alterations consisting of the combination of EGFR mutations with other EGFR mutations or with one or more mutations of other genes (tumour suppressor gene or oncogene). In some cases, only a small percentage of tumour cells has the specific EGFR mutation. The variable sensitivities to EGFR-TKIs could be explained by the variable tertiary structure of the EGFR protein under the influence of the different EGFR mutations [8].
1.1.1. Common EGFR mutations
Several studies reported that Del19 (45–50%) appears more frequently than L858R (37–40%) in Asian, as well as in Caucasian populations [4].
The Del19 and L858R mutations lead to elevated receptor dimerisation and activity due to the destabilisation of the inactive conformation of the EGFR receptor [9].
While the L858R mutation corresponds to the substitution of leucine to arginine at codon 858 (c.2573T > G, p.L858R), Del19 presents more than 30 variants, and the most-common is delE746_A750 corresponding to 73% of cases, with a deletion of 9 to 24 nucleotides [9]. In 25% of cases, Del19 variants start at position E747. The remaining percentage is known as entitled non-LRE (2%) [5][9][5,9].
1.1.2. Uncommon EGFR Mutations
Approximatively 600 EGFR uncommon or rare mutations have been described, accounting for 10–15% of cases, with variable sensitivity to EGFR-TKI treatment [10] and a similar clinical presentation in comparison with common ones. Examples of rare EGFR mutations are some EGFR exon point mutations such as exon 18, G719X; exon 20, S768I; and exon 21, L861Q [4][11][4,11].
1.1.3. Other EGFR Alterations
Complex mutations have a prevalence of 5–15% of all EGFR mutations. They are defined by the combination of common and rare EGFR mutations or rare and rare EGFR mutations or common and common ones. The most-frequent EGFR mutations involved in complex mutations are G179X (90%), G709X (75%), and S768I (50%) [12][13][14][15][12,13,14,15]. The sensitivity to EGFR-TKIs depends on the specific combination of mutations. It is better when one of them is a sensitivity mutation, such as Del19 or L858R, and lower when the combination includes a resistance mutation. For example, E709A + G719C, G787R + L858R, H870R + L858R, and E884K + L858R are sensitive complex mutations, while T790M + L858R is a resistant one [12][13][14][15][12,13,14,15].
Co-mutations correspond to the combination of EGFR mutation(s) with one or more mutations of another gene (tumour suppressor gene or oncogene) [16]. Co-mutations account for similar prevalence across the common EGFR mutations. Their incidence seems to be correlated with prior treatment. These genetic alterations often are found in several genes such as TP53, RB1, CTNNB1 (β-catenin), NKX2-1, or PI3KCA [17][18][19][17,18,19]. Some co-mutations are correlated with a worse prognosis; for example, TP53 mutations, ATM alterations, PTEN-inactivating mutations, KRAS mutations, and IDH1 mutations are associated with lower clinical results following EGFR-TKI treatment [17].
Subclonal mutations have a low variant allele frequency (VAF), which may be due to the presence of the specific mutation only in a small percentage of tumour cells [20]. All types of EGFR mutations could be subject to these genetic alterations, in particular the resistant ones. For example, a retrospective analysis of the AURA study, the AURA3 trial, and the study performed by the French Cooperative Thoracic Intergroup showed that the T790 mutation was present only in a small proportion of patients with worse clinical outcomes under third-generation EGFR-TKIs [21][22][23][24][21,22,23,24].
1.2. Clinical Trials
EGFR-TKIs became the standard therapy for advanced EGFR-mutation-positive NSCLC patients after the evaluation of their safety and efficacy in several clinical trials performed in the last decade. Moreover, some clinical studies documented a better prognosis, in terms of progression-free survival (PFS) and OS, for Del19 compared to L858R-mutation NSCLC patients under treatment with EGFR-TKIs [25][26][27][25,26,27].
1.2.1. First-Generation EGFR-TKIs: Gefitinib, Erlotinib, and Icotinib
The NEJ002 study compared gefitinib versus carboplatin plus paclitaxel as a first-line therapy for advanced NSCLC patients with a common EGFR mutation [28][29][28,29].
The IPASS trial investigated gefitinib with carboplatin plus paclitaxel in the same population of the NEJ002 study [30][31][30,31].
WJTOG3405 is a phase 3 study in which common-EGFR-mutation NSCLC patients were randomised between gefitinib and cisplatin plus docetaxel [32][33][32,33].
The OPTIMAL trial evaluated erlotinib with respect to standard chemotherapy as a first-line therapy for common-EGFR-mutation NSCLC patients [34][35][34,35].
The ENSURE study analysed erlotinib in comparison with gemcitabine plus cisplatin in Asian patients affected by common-EGFR-mutation NSCLC [36].
In the EURTAC trial, erlotinib was compared to standard chemotherapy as a first-line treatment for European patients affected by common-EGFR-mutation NSCLC [37].
The CONVINCE study was designed to evaluate the efficacy and safety of icotinib as a first-line therapy compared to cisplatin/pemetrexed plus pemetrexed maintenance for common-EGFR-mutation NSCLC patients [38].
All these trials reported a significant improvement in terms of PFS, but no statistical difference was seen for OS, maybe because of the high percentage of crossover from standard therapy to the experimental one after disease progression.
1.2.2. Second-Generation EGFR-TKIs
The LUX-Lung 3 trial evaluated afatinib versus cisplatin plus gemcitabine or pemetrexed for EGFR-mutation NSCLC patients stratified according to mutation type (exon 19 deletion, L858R, or other) [39].
In the LUX-Lung 6 study, common-EGFR-mutation NSCLC patients were randomised between afatinib versus cisplatin plus gemcitabine or pemetrexed [40].
ARCHER 1050 investigated the safety and efficacy of dacomitinib with respect to gefitinib as a first-line treatment of advanced NCSLC patients with a common EGFR mutation [41][46].
All these trials reported a significant improvement in terms of PFS, but no statistical difference was seen for OS, maybe because of the high percentage of crossover from standard therapy to the experimental one after disease progression.
1.2.3. Third-Generation EGFR-TKI
AURA3 was designed to evaluate the safety and efficacy of osimertinib in comparison with cis/carboplatin plus pemetrexed for advanced NSCLC patients who experienced disease progression after first-line EGFR-TKI therapy and developed the EGFR T790 mutation. Indeed, osimertinib is the third-generation TKI selective for T790M resistance mutations. The authors reported a longer PFS (10.1 versus 4.4 months; HR 0.30; p < 0.001) and OS (26.8 versus 22.5 months; HR 0.87, p = 0.277) for the osimertinib group, although the latter had no significant difference. This is probably due to the high crossover rate from chemotherapy to osimertinib of patients with progressive disease. After crossover adjustment, there was an HR of 0.54 for OS. The ORR was significantly better with osimertinib (71%) than the control group (31%) (odds ratio for OR: 5.39; p < 0.001). Encephalic PFS was also significantly longer for patients treated with osimertinib (8.5 months vs. 4.2 months; HR 0.32) [42].
The FLAURA trial tested osimertinib versus standard EGFR-TKIs (gefitinib or erlotinib) in previously untreated patients with common-EGFR-mutation NSCLC. The results reported PFS and OS significantly longer for the osimertinib group (PFS: 18.9 months vs. 10.2 months; HR 0.46; p < 0.001. OS: 38.6 months vs. 31.8 months; HR 0.80 p = 0.046). The ORR was similar (80% with osimertinib and 76% with standard EGFR-TKIs; odds ratio: 1.27; p = 0.24) [26]. As a consequence of the good results reported in this trial, osimertinib has become the first-line treatment for advanced or metastatic EGFR-mutant-positive NSCLC, regardless of T790M status.
The ADAURA study analysed osimertinib for 3 years as an adjuvant therapy for NSCLC patients with stage IB-IIIA and common EGFR mutations who have or have not previously received adjuvant chemotherapy. The DFS rate was 73% and 38% at 4 years (HR 0.27; p < 0.001) for the overall population in the osimertinib and control group, respectively. As regards CNS disease, at 24 months, 98% and 85% of patients in the experimental and placebo group were alive and did not have central nervous system disease (HR 0.18). The OS results are still immature [43].
1.2.4. EGFR-TKIs Specific for Ins20
ZENITH20-2 is a multicentre, multicohort, open-label phase 2 trial that investigated poziotinib for previously treated advanced NSCLC patients with EGFR exon 20 insertions that demonstrated resistance to approved TKIs. This type of mutation is an oncogenic driver and accounts for 2–5% of NSCLCs. The ORR was 27.8%. The disease control rate (DCR) was 70.0%, and PFS was 5.5 months [44].
Mobocertinib was tested in a phase 1/2, dose-escalation and dose-expansion trial that enrolled pretreated patients with advanced NSCLC and EGFR exon 20 insertions. This drug is a TKI targeting EGFR exon 20 insertions in NSCLC. The ORR was 43%, and PFS was 7.3 months [45].
2. Mechanisms of Resistance
Although all generations of EGFR-TKIs have been proven to be very effective for NSCLC with common EGFR mutations, almost 5–25% of these patients do not experience a clinical benefit with these drugs due to intrinsic resistance [46][47][57,58]. On the other hand, the major part of patients treated with EGFR-TKIs became resistant to these therapies despite an initial response or stable disease. The various mechanisms of resistance to EGFR-TKIs could be explained by the high molecular heterogeneity of NSCLCs (Figure 1 and Figure 2) [48][59]. Therefore, deepening the knowledge about the EGFR-TKI resistance mechanisms is one of the most-important aims in order to improve the treatment strategy of these patients.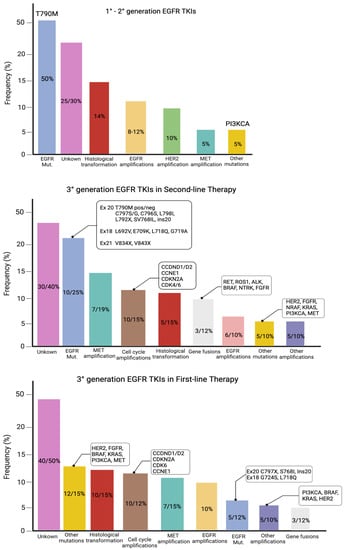
Figure 1. Mutations of resistance to EGFR-TKIs, according to the generation of TKIs and the line of therapy.
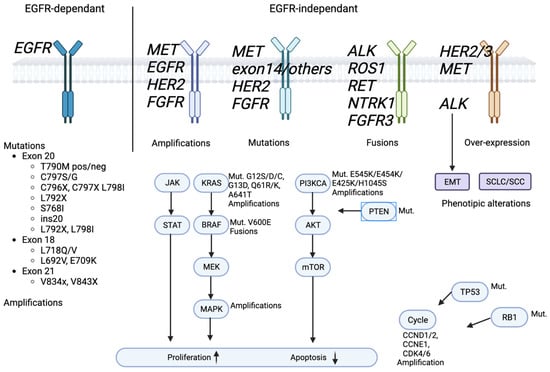
Figure 2. EGFR signaling pathway and EGFR-TKIs’ resistance mechanism.