(BEN)-Domain containing protein 3 (BEND3)D3 is a transcription factor that plays a critical role in the regulation of gene expression in mammals. While there is limited research on the role of BEND3 as a tumor suppressor or an oncogene and its potential role in cancer therapy is still emerging, several studies suggest that it may be involved in both the processes. Its interaction and regulation with multiple other factors via p21 have already been reported to play a significant role in cancer development, which serves as an indication of its potential role in oncogenesis. Its interaction with chromatin modifiers such as NuRD and NoRC and its role in the recruitment of polycomb repressive complex 2 (PRC2) are some of the additional events indicative of its potential role in cancer development.
- BEND3
- tumor suppressor
- oncogenic driver
1. Introduction
2. BEND3 Structure and Its Interaction with DNA
BEND3 is expressed from chromosome 6 in the human genome and has three introns and four exons. There are two reported transcriptional variants; nonetheless, both express the same functional protein. It has a long N-terminal loop followed by four BEN domains (BD1–4) of 80 amino acids each with distinct molecular interactions and functions. It is highly conserved across vertebrates. The nuclear localization signal (NLS) is present in the N-terminal loop. It forms an octameric higher-order structure [10]. Although the crystal structure of a full-length protein is not yet available, the structure of individual domains in mice (BD1, BD3, and BD4) [6] and humans (BD1 and BD4) [10][14][10,14] are available. The human BD1 domain is believed to be involved in the protein–protein interaction as it contains a coiled-coil domain and interacts with PICH1. The BD1 domain is exclusively composed of α helices with no beta turns [10]. The BD4 domain of BEND3 is composed of six α helices, two short helical turns, and two β strands. The DNA interaction region sits at the C-terminal and spans from α5 and α6. It is important to notice that both BD1 and BD4 domains share similar folds but perform distinct molecular functions. The BD1 domain lacks DNA binding activity [14]. The mouse BD3 domain shows similarity with the BD4 domain at the sequence as well as structural level, with few differences. The BD4 domain harbors DNA/chromatin binding activity. Unlike humans, mouse BD4 comprises five α helices, with the DNA binding module formed by α1 to α4 and the hinge region between α4 and α5 mediating dimer formation to accommodate two independent DNA molecules [14][15][14,15]. The NLS of this 95 KDa human protein consists of a Lysine-Arginine-Lysine motif [16] and, once in the nucleus, the BD4 domain can recognize and regulate its target genes. The BD4 domain is unique among other BEN domains as it has six α helices instead of five loops in others. In addition, it binds DNA through α5–loop–α6 sites while others bind through α5 and loop between α3 and α4 [14]. Zhang et al. has elaborately explained the crystal structure of the BD4 domain spanning from 715 to 828 in association with DNA [6].3. BEND3-Mediated Chromatin Regulation
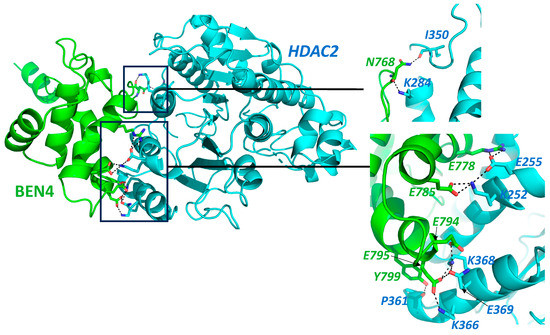
BEND3 interacts with various chromatin modifiers, including the subunits of the NuRD complex [8], suggesting its role in global gene regulation. The NuRD complex modulates the chromatin structure at the bivalent and poised rRNA genes [17] and various other genes in the ESCs [6][18][6,18]. It plays significant roles in processes like neural stem cell reprogramming into iPSC [19], pericentric heterochromatin formation [20], the maintenance of pluripotency [17][21][22][23][17,21,22,23], transcription repression [24][25][24,25], and S-phase progression [8]. The NuRD complex comprises various subunits such as CHD3/4, HDAC1/2, MBD2/3, MTA1/2/3, and retinoblastoma-binding protein (RBBP4/7). Other subunits such as LSD1 and GATAD2A/B have also been reported to associate with this complex in certain types of cells. Several subunits of the NuRD complex have implications in cancer development and progression. Its components, including MTA1, HDACs, Sal4, and CHD4, are expressed in different cancer types and corroborate the tumor progression and poor prognosis [11][26][11,26]. MTA1 is reported to be overexpressed in a wide range of cancer types and this overexpression correlates with tumor grade, poor prognosis, and invasion status of the tumor [11]. MTA1 acts as a downstream effector molecule of the Myc oncogene [27]. The NuRD complex is reported to interact with various oncogenic transcription factors and mediate the transcription repression of many target genes. In diffuse large B cell lymphoma (DLBCL), MTA3 associates with an oncogenic transcription repressor, BCL6 [28], which plays a significant role in the development of a significant proportion of DLBCL [29]. BCl6 requires MTA3 to transcriptionally repress normal plasma cell differentiation [28]. MTA proteins also interact with the transcriptional repressor BCL11B in leukemia and lymphoma cell lines. BCL11B has an indispensable role in early T cell development [11]. In breast cancer cells, a transcription factor, TWIST, causes the recruitment of MTA2 containing the NuRD complex at the CDH1 (E-Cadherin) promoter so as to repress E-Cadherin and promote the epithelial to mesenchymal transition (EMT), which is a hallmark of cancer, while MTA3 of the NuRD complex has been reported to cause transcription repression of SNAIL, thus inhibiting EMT in breast cancer cells. Thus, the complex can act as either a promoter or inhibitor of EMT depending upon the context of cells [11]. An oncogenic chimeric protein, PML-RARα, also recruits the NuRD complex, which in turn recruit other epigenetic regulators to cause transcriptional repression, leading to the impairment of cellular differentiation in human acute promyelocytic leukemias [30]. It is speculated that transcription factors recruit this complex to the promoters of the genes implicated in cancer [26]. Nine subunits of the NuRD complex are reported to be upregulated in hepatocellular carcinoma [26]. LSD1, part of the complex, regulates metastasis in breast carcinoma and is found to be under-expressed in breast cancers [31]. HDACs are important players in tumorigenesis and tumor progression and are overexpressed in several different kinds of malignancies including breast, colorectal, liver, pancreatic, ovarian, cervical, prostate, renal, bladder, melanoma, and certain blood cancers [32]. The BEN domain might act as an adapter in the process of chromatin modification by HDACs [15]. However, MTA1 and HDACs of the NuRD complex represent potential therapeutic targets for cancer chemoprevention. BEND3 also interacts with another NuRD complex transcription factor, Sal4, which is required to maintain the stemness and pluripotency of embryonic stem cells [9]. Sal4 expression is mis-regulated in various hematological as well as solid malignancies [33]. It plays a significant role in regulating various genes including apoptotic genes; cell-surface marker (EpCAM); EMT-related genes such as TWIST1, SNAI1, VIM, ZEB, E-Cadherin, etc.; and epigenetic modifiers such as DNMT1 and LSD1 [33]. Thus, it can be speculated that via its interaction with these subunits, BEND3 could aid in tumorigenesis mediated by the NuRD complex. Interestingly, BEND3 showed weak interaction with HDAC2 in our docking screen. The docking of BD4 was performed with HADDOCK 2.4 software and results are shown in Figure 1.