Idiopathic pulmonary fibrosis (IPF) is a disease that causes scarring and fibrotic transformation of the lung parenchyma, resulting in the progressive loss of respiratory function and, often, death. An increasing body of literature shows that pulmonary vascular permeability may play a big role in the pathogenesis of this condition. There is a search for therapeutic targets to try and modulate this vascular permeability in fibrotic lungs. One such class of targets that shows great promise is sphingolipids.
- idiopathic pulmonary fibrosis
- sphingolipids
- sphingosine-1-phosphate
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating disease, characterized by chronic scarring and fibrotic transformation of the lung parenchyma. It has a global prevalence of about 0.33–4.51 cases per 10,000 people after adjusting for age, sex, and smoking status. Patients with this condition suffer from debilitating dyspnea and cough, which is accompanied by a progressive decline in lung function, respiratory failure, and frequently, death. A meta-analysis of six global studies of IPF showed the three-year and five-year cumulative survival rates to be 61.8% and 45.6%, respectively [1]. These figures represent small improvements over past decades thanks in large part to current antifibrotic treatments. The two most important drugs in this category are pirfenidone and nintedanib.
Pirfenidone attenuates fibrosis by downregulating compounds such as transforming growth factor beta (TGF-β) and has reduced the rate of lung function decline for IPF patients in three phase III clinical trials [2][3][4][5]. Nintedanib is a tyrosine kinase inhibitor for receptors such as fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR). Like pirfenidone, it too has shown reductions in the rate of disease progression [3][6]. However, neither of these drugs has a major effect on mortality or is able to stop progression entirely; they also have many tolerability issues such as potent nausea and rash [3]. Thus, there is a need for the continued study of other pathways which influence the pathogenesis of IPF and therapeutics that modulate those pathways. Most previous research on IPF pathogenesis has focused on fibroblast activation and the epithelial-to-mesenchymal transition of the alveolar epithelium [7][8][9]. More recently, there is increased investigation into the role of vascular permeability in IPF pathogenesis. Increased vascular leak has been associated with fibrogenesis and areas of fibrosis have even been shown to colocalize with areas of increased capillary permeability in the lung [10][11]. This increased permeability may lead to greater extravasation of profibrotic and prothrombotic factors from blood vessels into the alveolar space, sustaining the process of fibrogenesis in fibrotic lungs. Consequently, examining pathways involved in vascular leakage and seeking ways to regulate those pathways can be the key to finding therapeutics that stall IPF progression.
Sphingolipids present a myriad of potential targets in this area. Sphingolipids are a class of nearly ubiquitous fatty acid derivatives of sphingosine that influence a variety of cell signaling pathways. Their numerous interactions with TGF-β and other profibrotic pathways have made them a subject of interest in the study of IPF pathogenesis [12][13][14]. Compounding research over the past decade shows that they also have numerous effects on vascular permeability. For example, in inflammatory conditions such as sepsis, ceramides can increase vascular permeability while S1P can promote endothelial barrier integrity and reduce vascular leakage [15][16]. This rule appears to apply in the lung vasculature as well. A series of papers have shown that the Sphingosine Kinase (Sphk)/S1P axis, especially through S1P receptor 1 (S1PR1), can reduce vascular permeability in the setting of pulmonary fibrosis, and thus, can attenuate the development of IPF [17][18][19]. ReIn this focusearchersd review we examine the current research around sphingolipids and the Sphk/S1P axis, and analyze how they affect endothelial barrier integrity, vascular permeability, and the development of IPF. ResearchersWe also discuss the possible benefits and drawbacks of trying to modulate this pathway to control vascular leakage in IPF patients.
2. Vascular Permeability in IPF Pathogenesis
In recent years, research has focused on the role that the pulmonary vascular endothelium plays in pathogenesis of IPF. In acute lung injury (ALI), increased vascular permeability is an essential component of the inflammatory response, allowing for the extravasation of immune cells into the ECM. This process is usually short-lived. However, a subject of current investigation is whether this permeability persists as tissue repair begins. Attempts at tissue repair after an acute injury will often result in fibrosis. Notably, studies have shown that the overexpression of pathways such as the Wnt/ β-catenin pathway, which promotes endothelial barrier integrity and reduces vascular leak, can attenuate the development of fibrosis in the days after an ALI. This implies that continued vascular permeability could correlate with increased formation of fibrotic tissue in conditions like IPF [20].
Indeed, research has shown that increases in vascular permeability are correlated with the formation of fibrotic foci and worse morbidity and mortality outcomes in IPF patients [10][11][21][22]. McKeown et al. in particular, found that higher indexes of permeability in IPF patients were associated with greater rates of rapidly declining lung function and death [10]. Thus, vascular leakage into the alveolar airspace could be a key sustaining factor of fibrogenesis in IPF patients, and medications that prevent this leakage could have major benefits for disease survivability [10][11].
3. Endothelial Barrier Permeability
To understand vascular permeability in lung disease processes, rwesearchers must first understand the endothelial cell layer. The vascular endothelial layer forms a barrier between the lumen of pulmonary blood vessels and the surrounding interstitial tissue. The integrity of this barrier plays a key role in regulating the flow of fluid and macromolecules between the vascular and interstitial space, and transport through this layer must be carefully controlled. Tight junctions and adherens junctions maintain this integrity [23]. Adherens junctions are composed primarily of vascular endothelial cadherins (VE-cadherin) that link to actin cytoskeleton [23]. Their main function is the forming of strong connections between the endothelial barrier cells [24]. Tight junctions, in contrast, are mostly composed of claudins and occludins and they work as a barrier on the apical side of the endothelium, regulating the passage of ions, water, and macromolecules [23][24].
At rest, the endothelial barrier stringently prevents plasma protein leakage and extravasation of leukocytes and other cells. In contrast, during inflammatory states, there is increased leakage of vascular contents into the interstitial space. The cells and plasma are often transported paracellularly, meaning they move through gaps formed between adjacent endothelial cells. There are numerous mechanisms by which paracellular gaps are opened; one important one is through increased actinomyosin contractility. The formation of actin stress fibers causes the contraction and shrinking of endothelial cells. This opens up paracellular gaps between the cells and increases permeability of the vascular bed [25][26].
Two cellular signaling pathways counteract each other to regulate endothelial barrier integrity via this mechanism: the Rho Kinase Pathway and the Rac Pathway. Rho associated coil kinases (ROCKs) are effector molecules of GTPases that increase the contraction of actin stress fibers [27]27; thus they increased paracellular gaps and vascular leak. On the other hand, the Rac pathway counteracts the actions of Rho by stabilizing the actin cytoskeleton and the inter-endothelial junctions, reducing vascular permeability [16][21][28] (16,21,28. (Figure 1). Molecular targets that can activate the Rac pathway and reduce this permeability could hold great potential as therapeutics for IPF. One such promising class of targets is sphingolipids [16][21][28]16,21,28.
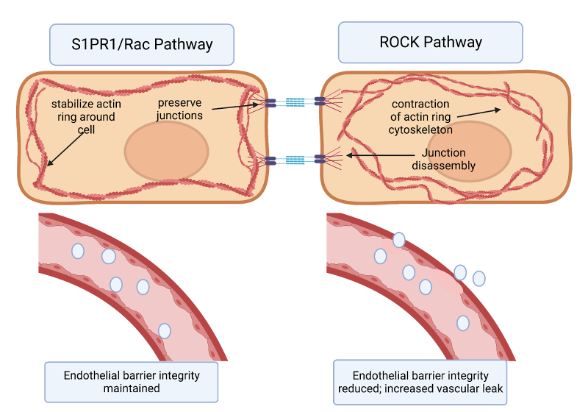
Figure 1. The effects of S1PR1/Rac pathway vs. ROCK pathway on endothelial barrier permeability. (a) Binding of the S1PR1 receptor activates the Rac pathway in endothelial cells. This primarily stabilizes the actin cytoskeleton and secondarily preserves endothelial junctions. In turn, endothelial barrier integrity is maintained. (b) Activation of the Rho kinase (ROCK) pathway primarily causes formation of actin stress fibers and circular contraction of the actin cytoskeleton, as well as cadherin junction disassembly. This contraction of actin cytoskeleton opens up paracellular gaps between endothelial cells, increasing vascular permeability 16,21,28,29.
4. Sphingolipids
Sphingolipids are a class of fatty acid derivatives of sphingosine that are present almost universally across eukaryotic cell membranes. Two particularly important and closely related sphingolipids are S1P and ceramide. The metabolic relationship between S1P and ceramide is best visualized by the sphingosine rheostat, a term first proposed in 1996 to describe the conceptual model of the relationship between these two molecules. In simple terms, it states that while ceramide is involved in apoptotic and growth-inhibiting pathways, S1P upregulates pathways involved in cell growth and inflammation (Figure 2). Ceramide can be converted into sphingosine by the enzyme ceramidase. Sphingosine can then be phosphorylated into sphingosine-1-phosphate by sphingosine kinase [14]14.
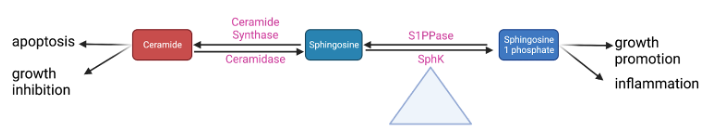
Figure 2. A simplified schematic of the sphingosine rheostat 14,30.
Some of these steps may play a larger role in regulating the whole pathway. Research from theour lab has shown that conversion between sphingomyelin and ceramide is rapidly reversible depending on whether the cell is in an inflammatory or quiescent state; as such, this point in the pathway could be important to maintaining equilibrium between different pro- and anti-inflammatory sphingolipids [30][31]30,31. Sphk is even more central to these pathways; it has been appropriately dubbed the “fulcrum” of the sphingosine rheostat as its activity controls the balance between S1P and ceramide levels (Figure 2) [32]32. There are two SphK isoforms: Sphk1 (the more predominant one) and Sphk2 [33]33. Despite having identical kinase domains, they have different properties, localize to different cellular areas, and likely have different functions. TheOur lab conducted early research into the differences between Sphk1 and Sphk2 and found that Sphk1 is significantly more involved in the upregulation of S1P [19]19.
There are five different membrane-bound G-protein-coupled receptors that S1P can bind to, and they are widely distributed across different cell lines (S1PR1-5). This resviearchw will focus on S1PR1, but researcherswe will briefly touch on the other receptors. S1PR2 is expressed in numerous organs and plays important roles in preventing apoptosis and enhancing growth. The activation of S1PR2 may play roles in both the preservation and disruption of endothelial barriers depending on the organ system [12][34][35]12,34,35 . S1PR3 is the second most common receptor on endothelial cells after S1PR1 [36]36. It is involved in many functions, but it is especially crucial to vascular development and the regulation of vascular tone [37][38]37,38. It also has immunomodulatory effects, but there is controversy over whether it is pro- or anti-inflammatory [12][34]12,34. S1PR4 is present mostly in hematopoietic tissues and affects lymphocyte signaling, as well as megakaryocyte and platelet activation. In the CNS it influences dendritic cell activation [34][39]34,39. S1PR5 is most heavily expressed in oligodendrocytes and other myelinating cells. Recent studies have shown that the activation of S1PR5 can be protective against conditions such as multiple sclerosis [34][40]34,40.
S1PR1 is the most studied of these receptors. It has numerous effects on cell lines throughout the body. Immune functions include the chemotaxis of lymphocytes from lymph nodes and proinflammatory signaling [14][34]14,34. Its function in the CNS is complex but includes promoting remyelination and astrocyte proliferation. There are currently FDA-approved S1P agonists for use in multiple sclerosis, and such drugs could play a major role in reducing sepsis-related encephalopathy [16]16. Lastly and crucially, S1PR1 is crucial to maintaining the integrity of the endothelial barrier in the vasculature [18][34]18,34.
5. The Effects of S1PR1 on Vascular Permeability
The link between sphingolipids and endothelial permeability in lung disease has been increasingly studied in the last decade. The idea was pioneered as far back as 2001 when researchers observed that the dose-dependent addition of S1P produced and sustained electrical resistance across layers of endothelial cells, indicating that S1P was strengthening the integrity of the endothelial barrier [41]41. Later, studies established links between S1PR1 and the Rac GTPase pathway, revealing that S1PR1 could reorganize peripheral actin rings, attenuating cellular contraction and preventing the formation of paracellular gaps (Figure 1) [42][43][44][45]42–45. Other research has shown that S1P signaling could affect VE-cadherin interactions, thus strengthening endothelial barrier integrity by manipulating inter-endothelial junctions [46][47]46,47.
RIn this researchersview, we previously discussed the potential links between vascular permeability and worsening fibrosis, morbidity, and mortality in IPF. ResearchersWe have also established that sphingolipids and S1P are tied to the regulation of endothelial vascular permeability. Consequently, it becomes crucial to examine whether modulation of sphingolipids such as S1P can affect vascular permeability in IPF, and if it has any effect on disease outcomes. Numerous previous studies have shown that the infusion of S1P into animal (usually murine or canine) models with acute or chronic lung injury reduces vascular leakage or leukocyte infiltration [19][48][49][50]48–51. These studies, however, did not delve into the specifics of how S1P might affect endothelial cells.
A series of papers by Knipe et al., published in 2018 and 2019, studied the role of S1PR1 and ROCK2 in endothelial cells in IPF [50][51]51,52. They induced endothelial-specific deletion of S1PR1 or ROCK2 in murine models and then induced IPF using a bleomycin challenge. By measuring dye extravasation and hydroxyproline levels, they were able to quantify vascular leakage and the development of pulmonary fibrosis, respectively. They found, as hypothesized, that the loss of S1PR1 resulted in increased vascular permeability and fibrosis while the deletion of ROCK2 was protective against these effects [50]51. This fits with theour existing model that the S1P/S1PR1 axis works in conjunction with the Rac pathway to stabilize cell–cell junctions and actin cytoskeletons, while the ROCK pathways increase actinomyosin contraction and open up paracellular gaps in the endothelial barrier (Figure 1) [21]21.
In 2020 and 2022, they used the same murine model with bleomycin-induced pulmonary fibrosis and focused on the results of endothelial-specific deletion of S1PR1. While continuing to measure vascular leakage and fibrosis, these papers also performed flow cytometry and measured D-dimer levels in both serum and bronchoalveolar lavage (BAL). The goal was to assess if S1PR1 knockout led to increased extravasation of clotting factors and inflammatory cells into the alveolar airspaces. Indeed, with the increased vascular permeability in S1PR1 knockout mice, researchers also observed increased coagulation factors and inflammatory cells in BAL samples, compared to controls [18]18. Furthermore, the 2022 study by Knipe et al. showed that even low-dose bleomycin was enough to cause a significant increase in fibrosis in S1PR1 knockouts. Histologic staining indicated that fibrotic areas co-localized with areas of increased vascular permeability, strengthening the link between these two factors [17]17.
Most interestingly, the above-mentioned paper noted that while the knockout mice had increased vascular permeability at baseline, they were phenotypically normal with no increased rates of fibrosis or mortality up to 6 months after S1PR1 deletion. This suggests that increased endothelial permeability does not cause fibrosis by itself, but perhaps strongly amplifies the effect of an inflammatory insult on the lung epithelium [17]17.
One possible way to conceptualize this is to view increased vascular permeability in S1PR1 knockout subjects as an opening for the extravasation of profibrotic and prothrombotic factors (Figure 3). As these factors leave the blood vessels, they enter the ECM and the alveolar airspaces where they can instigate tissue healing and eventual fibrotic transformation. For example, fibrin extravasation into the alveoli can lead to collagen deposition and maturation, potentiating more permanent fibrotic scarring of epithelial tissue [17]17]. Thrombin, while involved in coagulation, can also play a role in fibrogenesis. Direct inhibition of thrombin with dabigatran has been shown to reduce integrin αvβ6 induction and TGF-β activation, which then correlated with the reduced development of pulmonary fibrosis [52]53. Thus, in states of increased vascular permeability, such as in S1PR1 knockout mice, a greater leakage of prothrombotic factors into alveolar spaces can also induce fibrotic responses in alveolar epithelial cells [17][52]17,53. RWesearchers must note that Knipe et al. only knocked out S1PR1 in endothelial cell lines, and the effects they observed are only relevant to those cells [17]17. This is important because S1P is implicated in other
pathways that are potentially profibrotic.
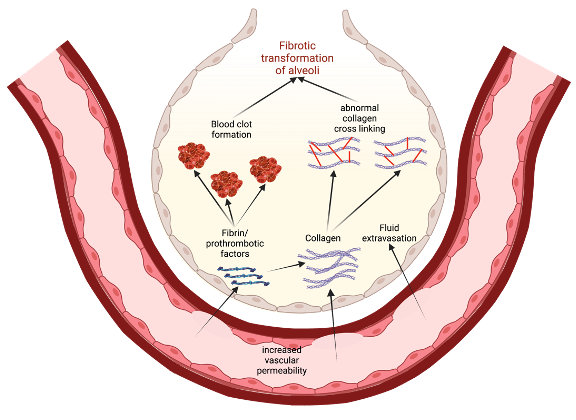
Figure 3. A model for how increased vascular permeability can potentiate fibrotic transformation of alveoli in IPF. This permeability allows for extravasation of prothrombotic factors and collagen that, in turn, promote clotting and activate profibrotic pathways. Eventually, there is recruitment of fibroblasts, abnormal collagen crosslinking, and a general epithelial-to-mesenchymal transformation in the alveoli, leading to lung fibrosis 17,53.
For example, Milara et al. found that S1P is increased in patients with IPF and actually promotes the epithelial–mesenchymal transition through potential crosstalk between the TGF-β1 and S1P/SPHK1 axis [53]54. Additionally, a study by Huang et al. found that in the alveolar epithelium, deleting Sphk1 and S1P reduces fibrosis by attenuating the Hippo/YAP pathway and decreasing TGF-β and mitochondrial reactive oxygen species activation [13]13. Because S1P could have profibrotic effects in epithelial cells and fibroprotective effects in endothelial cells, cell localization is something that should be considered if designing therapeutic targets for fibrosis based on sphingosine.
Finally, there is a small but important amount of research from the literature showing that S1PR1 may not be protective against vascular leakage and IPF development. The overexpression of S1PR1 can result in the receptor being internalized to the endoplasmic reticulum [54][55]55,56]. This action is performed with the help of chaperone proteins such as BiP/GRP78, and other research has shown that BiP/GRP78 can disrupt the endothelial barrier by other pathways that promote the disassembly of VE-cadherins and other endothelial cell–cell junction proteins [56]57. Thus, not only could the agonism of S1PR1 eventually lead to desensitization to its anti-permeability effects, but it could trigger other pathways that promote vascular permeability. Research by theour lab in 2009 showed that in a lipopolysaccharide induced (LPS) model of lung injury, the addition of S1P within six hours dramatically attenuated tissue damage and vascular leakage; however, the protective effects of S1P were no longer significant after six hours [19]48. A 2010 paper by Shea et al. supported these findings when trying to treat bleomycin-induced fibrosis with nonselective S1P1 agonists such as FTY 270 and AUY954. They found that in contrast to the protective effects of short-term exposure to these agonists, long-term exposure to these agents actually increased vascular leakage, fibrosis, and mortality [52]53. These results are consistent with theour understanding of S1PR1 internalization and emphasize the importance of considering the temporality of treatment when designing any IPF therapeutic based on sphingosine-1-phosphate.
6. The Effects of other S1P Receptors and Sphingolipids on Vascular Permeability
The bulk of the research and this review thus far have mainly focused on S1PR1. However, there are other S1P receptors to consider and other possible regulatory points in the Sphk/S1P axis. First, S1P can bind to five different receptors, and receptors one through three all have potentially different effects on vascular permeability and pulmonary fibrosis. There is very little research into the effects of S1PR2 on these outcomes. A 2007 paper by Sanchez et al. showed that activation of S1PR2 led to the increased activation of ROCK and phosphate and tensin homolog (PTEN) pathways and caused increased vascVular permeability, while the blockage of S1PR2 led to decreased permeability [36]36. In 2018, news studies showed that S1PR2 might also affect pulmonary fibrosis on a transcriptional level; S1PR2 deletion reduced the expression of profibrotic cytokines such as IL-13 and IL-4 in bleomycin-induced pulmonary fibrosis [57]58. Thus, it seems that S1PR2 might play the opposite role as S1PR1 in IPF, but the paucity of research makes any potential therapeutic role in IPF highly unclear.
S1PR3 also seems to have a profibrotic function. Studies have found that S1PR3 can promote epithelial-to-mesenchymal transformation and fibroblast activation in epithelial cells and ECM, respectively [58]59. Additionally, in endothelial cells, S1PR3 seems to promote increased endothelial barrier permeability. Murakami et al. found that S1P binding to S1PR3 can induce vasoconstriction and increased capillary permeability by activation of the ROCK pathway [12]12. Additionally, Sammani et al. found that S1PR3 knockout mice were protected against endothelial barrier disruption in IPF models [59]60. All of this indicates that S1PR3 probably plays an antagonistic role to S1PR1 in regulating endothelial barrier integrity.
Sphk can also play an important role by determining levels of S1P. As described previously, Sphk is the fulcrum of the rheostat, and though it is not as heavily studied in relation to IPF as S1P, it can still be an important regulatory point. TheOur lab demonstrated in 2009 that the knockout of Sphk1 exacerbated vascular leakage in an LPS-induced model of lung injury. ResearchersWe also found that the reintroduction of Sphk1 to endothelial cells via the adenovirus vector significantly attenuated permeability and lung damage [19]48. In contrast, researchwers found that the overexpression of Sphk2 in the Sphk1 knockout mice increased vascular leakage. While Sphk1 upregulates the production of S1P, SPhk2 seems to have a differing function, and this should be considered if trying to use sphingosine kinase as a therapeutic target [19]48. This function of Sphk1 in acute and subacute lung injury has largely been supported by reviews of the literature [58]59. However, there is conflicting research: Wang et al. showed that the upregulation of Sphk1 increased lung injury, while Sphk1 inhibitors attenuated vascular permeability. More research is needed to reconcile these differences in the literature [60]61.
7. Conclusions
A review of the literature reveals mounting evidence that increased vascular permeability may play an important role in the pathogenesis of IPF. A compelling conceptual model has been expressed in some of the literature to help explain this; increased permeability allows extravasation of profibrotic and prothrombotic factors into alveolar airspaces and triggers the transformation to fibrotic tissue . Consequently, modulating vascular permeability represents a promising therapeutic approach to treating IPF or stopping its progression. Sphingolipids represent one viable way to modulate that permeability. S1P/S1PR1 has been most heavily studied, and stimulation of S1PR1 can promote endothelial barrier integrity via interactions with Rac and other intracellular pathways. A growing body of research shows that S1PR1 can protect against vascular leakage and increased fibrosis in bleomycin models of IPF.
References
- Zheng Q, Cox IA, Campbell JA, et al. Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. ERJ Open Res. 2022;8(1):00591-02021. doi:10.1183/23120541.00591-2021
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33-41. doi:10.1016/S2213-2600(16)30326-5
- Spagnolo P, Kropski JA, Jones MG, et al. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. doi:10.1016/j.pharmthera.2020.107798
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet Lond Engl. 2011;377(9779):1760-1769. doi:10.1016/S0140-6736(11)60405-4
- Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821-829. doi:10.1183/09031936.00005209
- Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug Treatment of Idiopathic Pulmonary Fibrosis: Systematic Review and Network Meta-Analysis. Chest. 2016;149(3):756-766. doi:10.1016/j.chest.2015.11.013
- Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867-878. doi:10.1164/rccm.201403-0509PP
- Hewlett JC, Kropski JA, Blackwell TS. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol J Int Soc Matrix Biol. 2018;71-72:112-127. doi:10.1016/j.matbio.2018.03.021
- Yang J, Pan X, Wang L, Yu G. Alveolar cells under mechanical stressed niche: critical contributors to pulmonary fibrosis. Mol Med Camb Mass. 2020;26(1):95. doi:10.1186/s10020-020-00223-w
- McKeown S, Richter AG, O’Kane C, McAuley DF, Thickett DR. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J. 2009;33(1):77-84. doi:10.1183/09031936.00060708
- Declercq M, Treps L, Carmeliet P, Witters P. The role of endothelial cells in cystic fibrosis. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2019;18(6):752-761. doi:10.1016/j.jcf.2019.07.005
- Murakami A, Takasugi H, Ohnuma S, et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol Pharmacol. 2010;77(4):704-713. doi:10.1124/mol.109.061481
- Huang LS, Sudhadevi T, Fu P, et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int J Mol Sci. 2020;21(6):2064. doi:10.3390/ijms21062064
- Khan SA, Goliwas KF, Deshane JS. Sphingolipids in Lung Pathology in the Coronavirus Disease Era: A Review of Sphingolipid Involvement in the Pathogenesis of Lung Damage. Front Physiol. 2021;12:760638. doi:10.3389/fphys.2021.760638
- Jernigan PL, Makley AT, Hoehn RS, Edwards MJ, Pritts TA. The role of sphingolipids in endothelial barrier function. Biol Chem. 2015;396(6-7):681-691. doi:10.1515/hsz-2014-0305
- Kuperberg SJ, Wadgaonkar R. Sepsis-Associated Encephalopathy: The Blood-Brain Barrier and the Sphingolipid Rheostat. Front Immunol. 2017;8:597. doi:10.3389/fimmu.2017.00597
- Knipe RS, Spinney JJ, Abe EA, et al. Endothelial-Specific Loss of Sphingosine-1-Phosphate Receptor 1 Increases Vascular Permeability and Exacerbates Bleomycin-induced Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2022;66(1):38-52. doi:10.1165/rcmb.2020-0408OC
- Knipe RS, Spinney JJ, Abe E, et al. Loss of endothelial S1PR1 exacerbates bleomycin-induced pulmonary fibrosis through intra-alveolar coagulation and immune cell infiltration. Am J Respir Crit Care Med. 2020;201(1). https://www.embase.com/search/results?subaction=viewrecord&id=L632376616&from=export
- Wadgaonkar R, Patel V, Grinkina N, et al. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L603-613. doi:10.1152/ajplung.90357.2008
- Villar J, Zhang H, Slutsky AS. Lung Repair and Regeneration in ARDS: Role of PECAM1 and Wnt Signaling. Chest. 2019;155(3):587-594. doi:10.1016/j.chest.2018.10.022
- Probst CK, Montesi SB, Medoff BD, Shea BS, Knipe RS. Vascular permeability in the fibrotic lung. Eur Respir J. 2020;56(1):1900100. doi:10.1183/13993003.00100-2019
- Engelbrecht E, Kooistra T, Knipe RS. The Vasculature in Pulmonary Fibrosis. Curr Tissue Microenviron Rep. 2022;3(4):83-97. doi:10.1007/s43152-022-00040-9
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279-367. doi:10.1152/physrev.00012.2005
- Campbell HK, Maiers JL, DeMali KA. Interplay between tight junctions & adherens junctions. Exp Cell Res. 2017;358(1):39-44. doi:10.1016/j.yexcr.2017.03.061
- Wettschureck N, Strilic B, Offermanns S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol Rev. 2019;99(3):1467-1525. doi:10.1152/physrev.00037.2018
- Oldenburg J, de Rooij J. Mechanical control of the endothelial barrier. Cell Tissue Res. 2014;355(3):545-555. doi:10.1007/s00441-013-1792-6
- Katoh K, Kano Y, Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface. 2011;8(56):305-311. doi:10.1098/rsif.2010.0419
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39(4-5):187-199. doi:10.1016/s1537-1891(03)00008-9
- Del Gaudio I, Camerer E. Distinct GEFs Couple S1PR1 to Rac for Endothelial Barrier Enhancement and Lymphocyte Trafficking. Arterioscler Thromb Vasc Biol. 2022;42(7):903-905. doi:10.1161/ATVBAHA.122.317794
- Wadgaonkar R, Geraghty P, Kabir I, Foronjy R. Role of sphingomyelin synthase regulated micro domain signaling in cigarette smoke induced inflammation. Am J Respir Crit Care Med. 2017;195((Wadgaonkar R.; Geraghty P.; Kabir I.; Foronjy R.) SUNY Downstate Medical Center, Brooklyn, NY, United States). doi:10.1164/ajrccmconference.2017.C74
- Gowda S, Yeang C, Wadgaonkar S, et al. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L430-440. doi:10.1152/ajplung.00208.2010
- Wattenberg BW, Pitson SM, Raben DM. The sphingosine and diacylglycerol kinase superfamily of signaling kinases: localization as a key to signaling function. J Lipid Res. 2006;47(6):1128-1139. doi:10.1194/jlr.R600003-JLR200
- Siow DL, Anderson CD, Berdyshev EV, et al. Sphingosine kinase localization in the control of sphingolipid metabolism. Adv Enzyme Regul. 2011;51(1):229-244. doi:10.1016/j.advenzreg.2010.09.004
- Bravo GÁ, Cedeño RR, Casadevall MP, Ramió-Torrentà L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells. 2022;11(13):2058. doi:10.3390/cells11132058
- Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27(6):1312-1318. doi:10.1161/ATVBAHA.107.143735
- Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92(5):913-922. doi:10.1002/jcb.20127
- Forrest M, Sun SY, Hajdu R, et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309(2):758-768. doi:10.1124/jpet.103.062828
- Jin F, Hagemann N, Sun L, et al. High-density lipoprotein (HDL) promotes angiogenesis via S1P3-dependent VEGFR2 activation. Angiogenesis. 2018;21(2):381-394. doi:10.1007/s10456-018-9603-z
- Gräler MH, Grosse R, Kusch A, Kremmer E, Gudermann T, Lipp M. The sphingosine 1-phosphate receptor S1P4 regulates cell shape and motility via coupling to Gi and G12/13. J Cell Biochem. 2003;89(3):507-519. doi:10.1002/jcb.10537
- Niedernberg A, Scherer CR, Busch AE, Kostenis E. Comparative analysis of human and rat S1P(5) (edg8): differential expression profiles and sensitivities to antagonists. Biochem Pharmacol. 2002;64(8):1243-1250. doi:10.1016/s0006-2952(02)01289-3
- Garcia JG, Liu F, Verin AD, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108(5):689-701. doi:10.1172/JCI12450
- Berdyshev EV, Gorshkova I, Usatyuk P, et al. Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: role of sphingosine kinase 1 and S1P lyase. PloS One. 2011;6(1):e16571. doi:10.1371/journal.pone.0016571
- Adyshev DM, Moldobaeva NK, Elangovan VR, Garcia JGN, Dudek SM. Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal. 2011;23(12):2086-2096. doi:10.1016/j.cellsig.2011.08.003
- Donati C, Bruni P. Sphingosine 1-phosphate regulates cytoskeleton dynamics: implications in its biological response. Biochim Biophys Acta. 2006;1758(12):2037-2048. doi:10.1016/j.bbamem.2006.06.015
- Sun X, Shikata Y, Wang L, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77(3):304-313. doi:10.1016/j.mvr.2008.12.004
- Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology. 2011;76(8 Suppl 3):S3-8. doi:10.1212/WNL.0b013e31820d5ec1
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869-901. doi:10.1152/physrev.00035.2003
- Peng X, Hassoun PM, Sammani S, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169(11):1245-1251. doi:10.1164/rccm.200309-1258OC
- McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JGN. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med. 2004;170(9):987-993. doi:10.1164/rccm.200405-684OC
- Knipe RS, Spinney JJ, Abe E, et al. The pulmonary endothelium plays a critical role in the fibrotic response to lung injury through S1PR1 and rock mediated cytoskeletal rearrangements. Am J Respir Crit Care Med. 2019;199(9). https://www.embase.com/search/results?subaction=viewrecord&id=L630348969&from=export
- Knipe RS, Probst CK, Lagares D, et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2018;58(4):471-481. doi:10.1165/rcmb.2017-0075OC
- Shea BS, Probst CK, Brazee PL, et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight. 2017;2(9):e86608, 86608. doi:10.1172/jci.insight.86608
- Milara J, Navarro R, Juan G, et al. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax. 2012;67(2):147-156. doi:10.1136/thoraxjnl-2011-200026
- Brinkmann V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158(5):1173-1182. doi:10.1111/j.1476-5381.2009.00451.x
- Garnier O, Vilgrain I. Dialogue between VE-Cadherin and Sphingosine 1 Phosphate Receptor1 (S1PR1) for Protecting Endothelial Functions. Int J Mol Sci. 2023;24(4):4018. doi:10.3390/ijms24044018
- Leonard A, Grose V, Paton AW, et al. Selective Inactivation of Intracellular BiP/GRP78 Attenuates Endothelial Inflammation and Permeability in Acute Lung Injury. Sci Rep. 2019;9(1):2096. doi:10.1038/s41598-018-38312-w
- Zhao J, Okamoto Y, Asano Y, et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PloS One. 2018;13(5):e0197604. doi:10.1371/journal.pone.0197604
- Natarajan V, Dudek SM, Jacobson JR, et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol. 2013;49(1):6-17. doi:10.1165/rcmb.2012-0411TR
- Sammani S, Moreno-Vinasco L, Mirzapoiazova T, et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43(4):394-402. doi:10.1165/rcmb.2009-0223OC
- Wang Y, Gao TT, Xu DF, et al. Upregulation of sphingosine kinase 1 contributes to ventilator-associated lung injury in a two-hit model. Int J Mol Med. 2019;44(6):2077-2090. doi:10.3892/ijmm.2019.4379