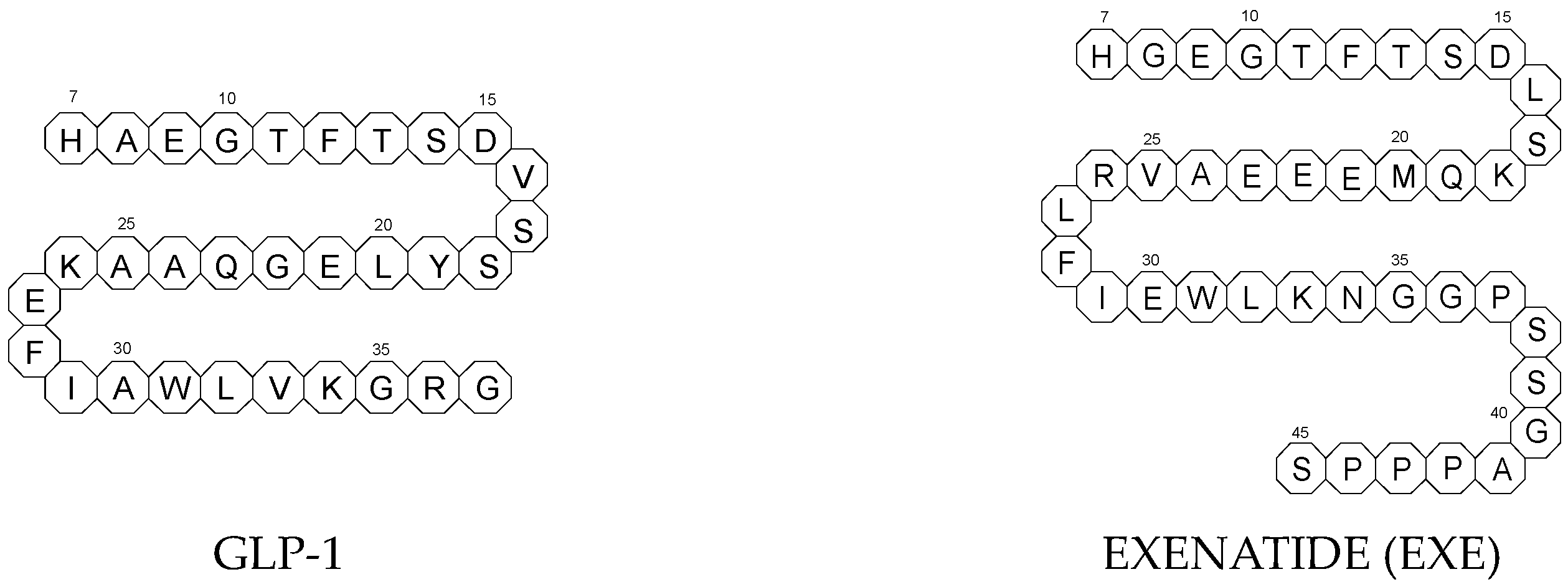The drug metabolism and drug degradation pathways may overlap, resulting in the formation of similar constituents. Therefore, the metabolism data can be helpful for deriving safe levels of degradation impurities and improving the quality of respective pharmaceutical products. The prentrysent article contains considerations on possible links between metabolic and degradation pathways for new antidiabetic drugs such as glutides, gliflozins, and gliptins. Special attention was paid to their reported metabolites and identified degradation products. At the same time, many interesting analytical approaches to conducting metabolism as well as degradation experiments were mentioned, including chromatographic methods and radioactive labeling of the drugs.
- drug metabolism and drug degradation
- chromatographic and radiometric methods
- in vitro metabolic studies
- peptide drugs
- glutides and gliptins
1. Introduction
2. Analytical Tools for Peptide Drugs Examination
The best methods for the separation of peptide products include HPLC, UPLC, ion exchange high performance liquid chromatography (HPLC-IEX), and size exclusion high-performance liquid chromatography (HPLC-SEC). Furthermore, the high sensitivity and selectivity of MS coupled with LC or UPLC (HRMS) are suitable for the identification of peptides, their metabolites, and degradation products. For peptide compounds, the most widely used ionization modes in MS are electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) techniques [11][15]. Mass spectrometers can also be combined with a quadrupole, such as a Q-TOF system, in which fragmentation can increase the selectivity of these methods. Furthermore, ion trap and orbitrap mass analyzers that deliver excellent resolution and mass accuracy could be applied. These accurate HRMS systems detect a wide range of compounds as well as small molecules during both targeted and untargeted analyses without losing selectivity or sensitivity. Due to its high specificity, HRMS allows measuring the majority of peptides, including those with changed sequences, even if they are not completely separated. Even when changes to the primary amino acid sequence in peptides due to degradation or other changes occur, they could be recorded as detectable mass shifts. Furthermore, metabolomics studies of peptide drugs are frequently performed using HRMS [10][11][11,15]. Generally, peptides can be separated with the most frequently used C18 columns; however, some of them can give poor peak shape or peak tailing. For these reasons, C4 columns are recommended for the separation of peptides. Furthermore, phenyl columns, which are similar to C4 columns as far as their hydrophobicity is concerned, could be used for peptide analysis. In the literature, the usefulness of polar embedded or polar endcapped columns, in which polar interactions between peptides and the particle surface are enhanced, is also reported. In addition, peptide drugs could be more effectively separated using longer columns than those usually used in the separation of small drug molecules. Thus, columns with a length of 15 or 25 cm are recommended. As far as mobile phases are concerned, the reversed phase (RP) mode of chromatography could be used for peptides, but it usually requires the use of ion-pair reagents in order to achieve a good peak shape for the analytes [12][16]. Among the best-rated ion pair reagents for such analysis is trifluoroacetic acid (TFA), which can be added to the mobile phase at a concentration of 0.1%. In consequence, the mobile phases used for peptide separations in RP systems and containing TFA are generally adjusted to a low pH. At such conditions, the carboxylic groups of amine acids are non-ionized and only slightly polar. On the other hand, when the pH of the mobile phase rises to 6–7, the carboxylic groups tend to ionize, making the peptide less hydrophobic. To carry out a forced degradation study of macromolecules, generally, temperature, pH change, and agitation are used as factors to check the instability of the desired molecules. However, small peptides should be subjected to acidic, basic, and neutral hydrolysis, oxidation, photostability, and thermal treatment, according to the strategy of ICH guidelines Q1A(R2) [13][17], just like the small molecule drugs. At the same time, the selection of appropriate stress conditions is essential and constitutes the most crucial step in conducting the decisive forced degradation study. It is recommended to optimize the stress conditions in such a way to attain degradation kinetics thermodynamically equivalent to accelerated stability conditions [14][18]. It is known that excessive stress can lead to the secondary degradation of products.3. Glutides (GLP-1 RAs)
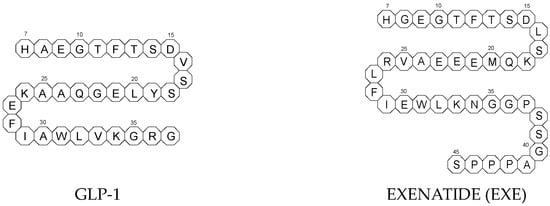
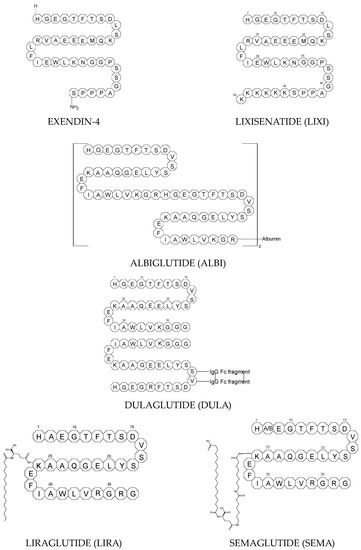
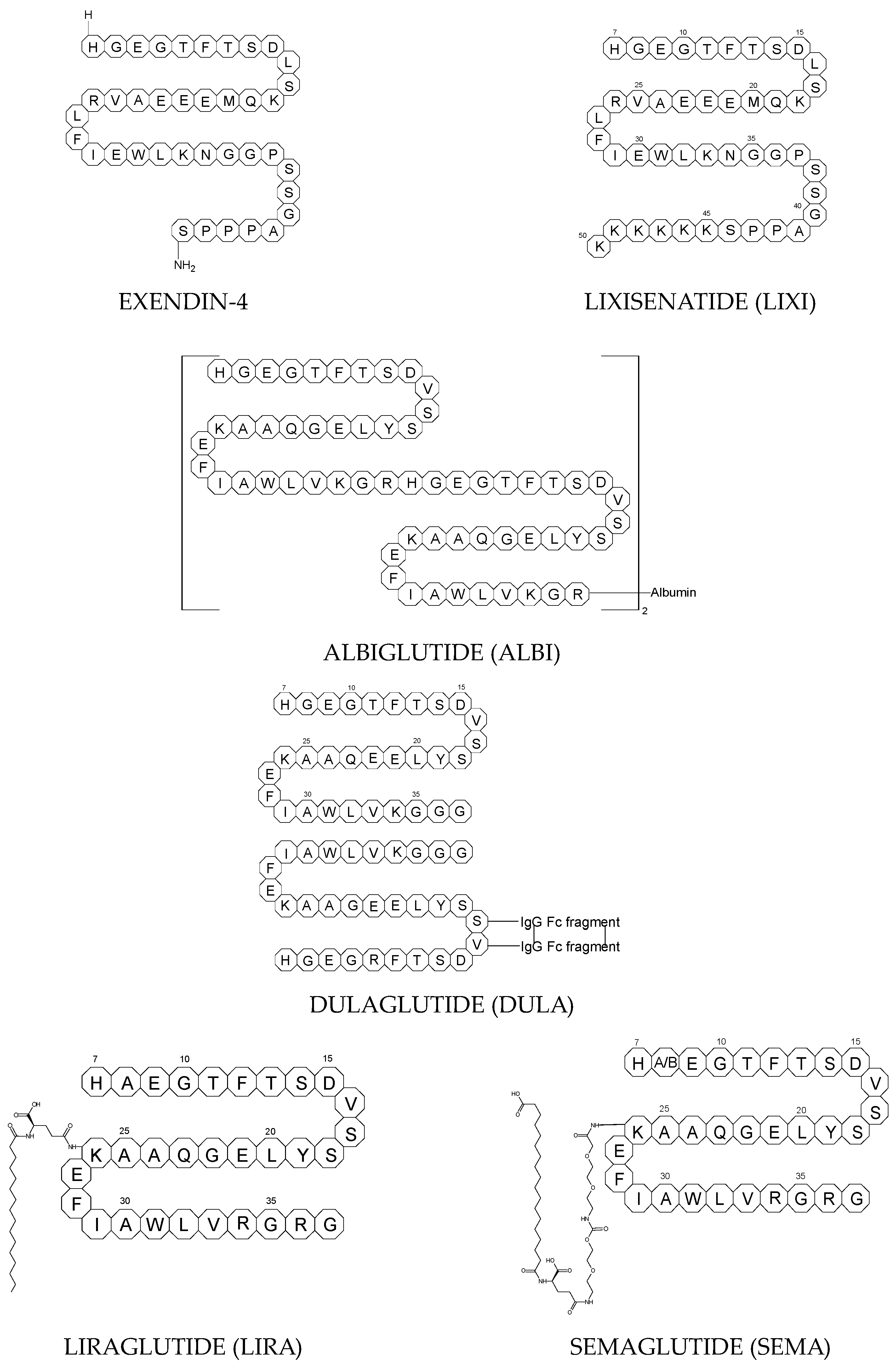
To ameliorate the altered incretin effects in T2DM, glutides or the GLP-1 RAs, i.e., albiglutide (ALBI), dulaglutide (DULA), exenatide (EXE), liraglutide (LIRA), lixisenatide (LIXI) in injectable formulations, and semaglutide (SEMA) in injectable and oral formulations, are introduced as effective therapeutic approaches. It is well known that the incretin effect contributes nearly half of the insulin secretory response to oral glucose load and that this effect is reduced along with worsening glucose tolerance in diabetic patients. The glucose-dependent insulinotropic polypeptide, formerly known as gastric inhibitory peptide (GIP), and glucagon-like peptide-1 (GLP-1) are the two primary incretins secreted from the intestine on ingestion of glucose to stimulate insulin secretion from pancreatic b-cells, enhance satiety, and delay gastric emptying. GIP and GLP-1 exert their effects by binding to their specific receptors, the GIP receptor and the GLP-1 receptor (GLP-1 R), which both belong to the G-protein-coupled receptor family. Physiologically, GIP and GLP-1 are rapidly degraded by the enzyme dipeptidyl peptidase 4 (DPP-4), which acts on peptides to cleave the two NH2-terminal amino acids [15][16][20,21]. Thus, GIP and GLP-1 share common properties as incretins, but they also possess different biological characteristics. Endogenous GIP exerts strong insulinotropic effects in healthy subjects, but its insulinotropic effect is seriously reduced in diabetic patients, which discourages the development of GIP-based therapies. In contrast, the insulinotropic effect of GLP-1 is preserved in T2DM patients, establishing GLP-1 and GLP-1R signaling as attractive therapeutic targets [16][21]. GLP-1 RAs exert a dual action on the endocrine pancreas cells, with a stimulation of insulin secretion by b-cells, mainly in the postprandial state, and an inhibition of glucagon secretion by a-cells, contributing to reducing hyperglycemia. In addition, they slow down gastric emptying and activate the hindbrain GPL-1 receptors. All glutides present on the market are synthetic peptides with high homology with human GLP-1 (7–37). EXE, which is given to patients subcutaneously twice daily, was the first GLP-1 RA introduced into therapy in 2005 (FDA approval). It is a 39-amino acid synthetic peptide with 53% homology with human GLP-1 (7–37). It contains an Ala8Gly substitution that increases its resistance to degradation by DPP-4. Next GLP-1 RAs, including subcutaneously once daily administered LIRA and LIXI and once weekly given ALBI and DULA, were approved by FDA in 2021, 2016, and 2014, respectively. LIRA is a true analogue of GLP-1 (97% homology) with the addition of a 16-carbon palmitic acid chain conjugated via a glutamate spacer at Lys in position 26, to mask the DPP-4 cleavage site. LIXI is an exendin-4 analogue (94% sequence homology with GLP-1) with 6 Lys residues added to allow resistance to DPP-4. ALBI consists of 2 copies of GLP-1, each with an Ala8Gly substitution, and as a whole, the molecule is fused to albumin [17][23]. DULA has two copies of a GLP-1 analogue (with amino acid substitutions Ala8Gly, Gly22Glu, and Arg36Gly) that are covalently linked to an Fc fragment of human IgG4 [18][24]. The next GLP-1 RA, i.e., SEMA, is marketed as a once-weekly subcutaneous injection and was approved by the FDA in 2017. It is an analogue of LIRA with a substitution of Ala at position 8 with an aminoisobutyric acid (A/b). The C16 fatty acid is also exchanged for the C18 fatty acid and linked by a synthetic spacer. Acylation with a spacer and C-18 fatty acid chain increases its binding to blood albumin, which enables its longer presence in the blood circulation. Despite the above-mentioned subcutaneous form of SEMA, its orally administered analog with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl]amino)caprylate (SNAC) to overcome the problems of poor absorption and degradation in the stomach was approved in 2019 [16][19][21,25]. The structures of the mentioned GLP-1 RAs are presented in Figure 1.