Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giuseppe Murdaca and Version 2 by Peter Tang.
Multiple Myeloma (MM) is a haematological disease resulting from the neoplastic transformation of plasma cells. The uncontrolled growth of plasma cells in the bone marrow and the delivery of several cytokines causes bone erosion that often does not regress, even in the event of disease remission. MM is characterised by a multi-step evolutionary path, which starts with an early asymptomatic stage defined as monoclonal gammopathy of undetermined significance (MGUS) evolving to overt disease.
- alarmins
- cytokines
- multiple myeloma
1. Introduction
Multiple Myeloma (MM) is a haematological disease resulting from the neoplastic transformation of plasma cells, which are the terminally differentiated cells of the B lymphocyte line [1][2][1,2]. Malignant plasma cells can invade not only primarily the bone marrow but also peripheral blood and viscera in advanced widespread [3]. Plasma cells physiologically produce immunoglobulin (Ig). Malignant plasma cells produce and release excess monoclonal protein (M protein) in serum, which is also known as paraprotein [4]. The M protein will be excreted in the urine. Numerous genetic alterations favor the onset of the uncontrolled proliferation of plasma cells and the production of paraprotenin [1][2][1,2]. The uncontrolled growth of plasma cells in the bone marrow and the production of cytokines cause bone erosion, and the resulting bone lesions often do not regress, even in the event of disease remission. MM is the second most common haematological malignancy, and it is characterised by the appearance of bone pain, hypercalcemia, anemia, and renal failure [1][2][3][1,2,3]. MM is characterised by a multi-step evolutionary path, which starts with an early asymptomatic stage defined as monoclonal gammopathy of undetermined significance (MGUS) evolving to overt disease. MGUS is classified according to the secretion of Ig and, thus, into MGUS IgM and MGUS non-IgM. Although MGUS in most cases has a benign course, in some cases, it can evolve into aggressive forms. In particular, IgM MGUS can develop in Waldenström macroglobulinemia (WM) or, in fewer cases, in other non-Hodgkin’s lymphomas, while non-IgM MGUS originating from mature plasma cells can develop in MM [5]. MGUS can also secrete only the κ or λ light chain of Ig. Light chains aggregating are the cause of organ damage (i.e., heart, kidneys) both by depositing themselves or by favoring the deposition of amyloid from light chains (AL) [6][7][6,7]. However, in patients with MGUS, the M protein is <30 g/L and represents 20%–70% of all Ig, and circulating monoclonal plasma cells are <3 × 106 per liter [8]. The advent of new drugs such as proteasome inhibitors and immunomodulatory agents have certainly improved the prognosis and median overall survival of MM to over 60 months [9]. However, MM still remains an incurable disease. The clinical picture and its progression over time is favored and aggravated by the inevitable onset of drug resistance [10][11][12][13][10,11,12,13]. Plasma cell dysfunction and uncontrolled proliferation, granulocytopenia both from tumour marrow invasion and iatrogenic from chemotherapy, and high-dose administration of dexamethasone promote immunodeficiency in the patient with MM. Immunodeficiency favors both recurrent opportunistic infections and the evasion of the tumour from the immune response with its progression and wide spread [10][14][15][16][17][10,14,15,16,17]. Immunodeficiency is the consequence of both the weakening of B- and T-lymphocyte response, but also the antigen-presenting cells (APC); the natural killer (NK) cells are compromised in their number and functionality [10][18][19][20][21][22][23][24][10,18,19,20,21,22,23,24].
The previously reported alterations of the effectors of the immune system at the bone marrow level seem fundamental not only in the determinism of the disease but also in its progression. In MM, the crucial function of the BM tumour microenvironment and in particular in the effectors of the immune system contained therein is well recognised, and numerous reports have reported that plasma cells intensely depend on it [25]. MM plasma cells cooperate, directly and indirectly, with the bone marrow milieu stimulating growth, survival, and chemoresistance [26].
Moreover, a recent investigation revealed that in the BM, MM mesenchymal stem cells (MSCs) have a specific gene expression profile with respect to the normal MSCs. The evaluation of the gene co-expression network demonstrated that the principal altered activities in MM-MSCs are correlated to cell cycle progression and modifications of the immune response, which may participate in MM physiopathology [27].
Furthermore, several findings demonstrated that tumour progression in MM is linked to a change of tumour-specific immunity [28], proposing that immune surveillance may have an action in the prevention of MM disease evolution, as communications with the contiguous milieu are essential to MM cells’ survival [29]. BM immune effector interactions with plasma cells are essential for MM evolution, as contacts increase the generation of different anti-apoptotic and cell cycle-stimulating factors [30]. Plasma cells use numerous mechanisms to escape immune surveillance, which comprise modified interactions with T-cells, dendritic cells, and natural killer cells. These changes can be due to immunosuppressive cells and cytokines such as IL-10, TGFβ, and IL-6 as well as a reduction of the antigen processing machinery [31].
2. Role of the Immune System in MM Progression
The presence of T cells specific for the idiotype of the monoclonal paraprotein is described in patients with MGUS and MM, although there is still no definitive evidence on their actual role [32][33][90,91]. IL-1 plays a pivotal role in the progression from MGUS to MM as well as having a proinflammatory role. It is now well established that the plasma cells of patients with MM exhibit high concentrations of IL-1β. On the contrary, IL-1β is not detectable in the plasma cells of patients with MGUS. Therefore, the productive switch of IL-1β would represent a signal of malignant progression of the disease [34][35][92,93]. Confirming its probable role in the progression from MGUS to MM, IL-1β would seem to enhance the ability of plasma cells to cause lytic bone lesions, to induce the expression of adhesion molecules such as ICAM, CD44, CD54, CD56, CD44, and VLA-4 on plasma cells by increasing their capacity for transmigration and metastatic widespread [36][37][38][39][40][94,95,96,97,98]. It is hypothesised that a viral infection may represent the initial stimulus capable of inducing the upregulation of IL-1β. Among the viruses involved, there would be Kaposi’s sarcoma-associated herpesvirus, Epstein–Barr virus, human immunodeficiency virus-1, and respiratory syncytial virus [41][42][99,100]. An interesting finding that could give a further potential explanation of the cytokine network that favors disease progression would be related to the increased expression of IL-2R on MM cells and to a lesser extent on MGUS cells, suggesting that progression from MGUS to MM is also related to the alteration of the IL-2/IL-2R system [43][101].3. Neoangiogenesis in MM
The elevated expression of HMGB1 has an undoubted role in the transformation of MGUS into MM and in the progression of MM itself, blocking cell apoptosis and inducing drug resistance by regulating the NF-κB signalling pathway. Notably, HMGB1 induces neoangiogenesis in patients with solid tumours and other hematological neoplasms. Meyer et al. [44][102] demonstrated in 18 non-Hodgkin lymphomas and two lymphoma cell lines that HMGB1 probably released from necrotic cells promotes tumour neoangiogenesis in a paracrine way. Zhan et al. [45][103] confirmed that autophagy induced the release of HMGB1 by gastric cancer cells into the extracellular space after exposure to vincristine, protecting gastric cancer cells from apoptosis through the upregulation of Mcl-1. Shrivastava et al. [46][104] demonstrated that elevated levels of HMGB1 reduced the response to radiotherapy in eight human urothelial carcinoma cell lines. Zhang et al. [47][105] confirmed HMGB1 expression in non-small cell lung cancer cell lines and that exposure to adriamycin, cisplatin, and methotrexate further increased HMGB1 levels. However, Chen et al. [48][106] confirmed the ability of HMGB1 to protect chronic myeloid leukemia cells from apoptosis, as proven in other studies [49][50][51][52][107,108,109,110]. Wu et al. [53][111] analysed the impact of HMGB1 expression on clinical progression in 166 patients with nasopharyngeal carcinoma. The study showed that the high expression of HMGB1 increased the risk of metastatic disease with poor prognosis. Süren et al. [54][112] confirmed the negative impact of HMGB1 in 110 patients with colorectal carcinoma. Several studies have shown that HMGB1 blockade restores apoptosis. It is plausible that HMGB1 promotes neoangiogenesis through the expression of vascular endothelial growth factor (VEGF) as occurs in chronic immune-mediated diseases and allergies such as allergic rhinitis [55][56][113,114]. Ostecytes play a key role in the progression of MM by promoting neoangiogenesis through the expression of molecules, including VEGF [57][115]. It is now shown that VEGF by stimulating its type 2 receptor (VEGFR2) induces neoangiogenesis, favoring the progression of MM [58][116]. Single nucleotide polymorphisms (SNPs) of VEGF and VEGFR2 genes have been demonstrated to play a key role in MM progression. For example, the presence of the VEGFR2-604TT genotype is associated with stage II or III tumours but not with stage I tumours [58][116]. Furthermore, endothelial dysfunction that accompanies neoangiogenesis in turn favors the passage of immune cells into the tumour environment through the expression of direct chemokines and cell adhesion molecules on the surface of endothelial cells as also occurs in chronic immune-mediated diseases [59][60][117,118].4. Drug Resistance in MM
Crosstalk between adaptive immune cells and the endothelium is critical to tumour immune surveillance and the success of immune-based therapies that use immune cells to kill tumour cells. There are several factors that can reduce the pharmacological response of MM, resulting in disease progression. Notably, MicroRNAs (miRNAs) are an abundant group of endogenous non-coding RNAs (about 22 nt) binding to target mRNAs mainly at their 3′-untranslated (UTR) that displayed a key role in lung cancer [61][119]. However, miR-218 can inhibit the metastatic spread of tumours including non-small cell lung cancer and pancreatic cancer by blocking the expression of HMGB1 [62][63][120,121]. However, miRNAs appear to play a role in resistance to chemotherapy drugs. Ran et al. [64][122] demonstrated that the downregulation of miRNAs is associated with resistance to paclitaxel in endometrial cancer cells. Notably, miR-218 binds directly to the 3’-UTR of the HMGB1 gene, the upregulation of which induces resistance to paclitaxel in endometrial cancer cells by promoting autophagy. MiR-218 overexpression would inhibit HMGB1-induced autophagy by restoring tumour cell response to paclitaxel.5. MGUS to MM: Role of Immunoglobulin Chains
The roles that the quantity and type of M protein and the ratio of free immunoglobulin chains have in the evolution of the disease from MGUS to MM is now widely accredited. It is also evident that the suppression of the production of PIg is not only a progression factor but also has a negative impact on the clinical evolution favoring “immune paresis” and consequently increasing the risk of opportunistic infections [65][66][33,68]. Rapid progression from MGUS to MM is favored by the decreased synthesis of IgM in subjects with IgG or IgA gammopathy, and of IgA in individuals with IgG or IgM gammopathy [67][68][69][123,124,125]. The network that influences immune paresis is certainly complex, and it is not yet completely clear whether it all starts from the dysregulation of cytokines and whether this dysregulation is due to defects in immune surveillance, the proliferation of clonal plasma cells, or opportunistic infections [70][126]. Moreover, the immune paresis reaches its peak with old age but decreases among the very elderly [71][127]. HMGB1 facilitates autophagy by disrupting the interaction between Beclin 1 and its downregulator Bcl2 via competitively binding to Beclin 1 [72][128]. Wang et al. confirmed that increased HMGB1 expression in the neuroblastoma NB SH-SY5Y cell line induced resistance to treatment with doxorubicin, etoposide, and cisplatin. Sensitivity to treatment was restored after HMGB1 was knocked out by RNA interference [54][112]. HMGB1 has been shown to induce elevated levels of LC3-II (a protein light chain associated with microtubules that plays a role in autophagosome formation) and the selective degradation of p62 (a ubiquitin-related protein) in autophagy. In this way, the green fluorescent protein-light chain 3 favors the generation of autophagosomes [54][112].6. HMGB1/RAGE Axis as Crosstalking between Immune Cells and Bone Tissue
In patients with pancreatic cancer, the mechanism of action of HMGB1 is not yet fully understood, although it seems to have a dual action. It is likely to inhibit tumourigenesis by stabilising the genome through its direct binding to p53 at the intracellular level and by binding to certain receptors including RAGE, TLR-2, TLR-4, and TLR-9 as well as by interacting with cytokines and growth factors at the extracellular level [55][113]. In patients with MM, a plant-based chemical lycorine has been shown to be able to decrease HMGB1 and, therefore, LC3 and Beclin-1, thereby blocking autophagy. In particular, lycorin dissociates Bcl2 from Beclin-1 by acting through the ubiquitin–proteasome pathway [72][128]. Lycorin restores the response of MM patients to bortezomib treatment by reducing HMGB1 expression. These data confirm that lycorine has a dual activity: both antitumourigenic and promoting sensitisation to chemotherapy in MM [73][129]. There is no doubt that the not too distant future approach will be to design and synthesise monoclonal antibodies directed against specific molecules including HMGB1 to make therapies targeted and hopefully with fewer adverse effects [74][130]. Confirming this, Usman et al. [75][131] have reported some clinical trials targeting HMGB1. RAGE is among the receptors to which HMGB1 binds, and it is a member of the immunoglobulin superfamily and is a transmembrane receptor that binds to AGE. HMGB1 activates RAGE or activates the peroxisome via the proliferator-activated receptor gamma (PPAR-γ) with consequent inhibition of the HMGB1-RAGE axis, and this mechanism could also represent a future therapeutic strategy beneficial against tumours [76][77][73,74]. Furthermore, CXCR4 expression is ubiquitous in different hematopoietic cells [78][132], and its role in the evolution of MM has already been demonstrated [79][133]. It is now well established that elevated levels of CXCR4, integrins (i.e., CD11a/CD11c/CD29/CD49d/CD49e), and adhesion molecules (i.e., CD44/CD54) in patients with MM induce resistance to chemotherapy [80][81][134,135]. Furthermore, the HMGB1/RAGE axis has now been shown to be able to favor bone loss. HMGB1 is expressed in primary osteoblasts and osteoclasts both expressing RAGE. Furthermore, the RANKL/osteoprotegerin (OPG)/receptor activator of NF-κB (RANK) signalling axis plays a critical role in DCs function as well as bone remodeling. RANKL is present in DCs that release HMGB1 and represents a mode of crosstalking between immune cells and bone tissue. These data would confirm the complexity of the mechanisms and the role of the bone marrow microenvironment in the progression of MM [82][83][136,137]. RAGE is encoded in the Class III region of the major histocompatibility complex and it is the multiligand receptor of the immunoglobulin superfamily. RAGE stimulated by AGEs has been shown to trigger pro-tumourigenic pathways by promoting the dissemination of melanoma and pancreatic cancer cells [83][137] and probably has a similar action also in colorectal, oesophageal, and oral squamous cell carcinoma, since in these tumours, it is upregulated and, therefore, acts as an oncoprotein [55][56][113,114]. The fact that levels of sRAGE are higher in the IgA Lambda MM, which has a poorer prognosis, is explained as a reactive mechanism to the tumour, which is fully confirmed by the ability of the same sRAGE to bind other ligands such as proinflammatory molecules. There is no doubt that HSPs also play a role in the activation of proinflammatory cytokines secreted by monocytes/macrophages and favor the activation of immature DCs [60][84][65,118]. IL6 increases the expression of HSP90 in patients with MM, thus favoring the survival of myelomatous plasma cells. It is now shown that the increase in HSP90 levels is induced by STAT3 and CCAAT/enhancer-binding protein β (C⁄EBPβ), which bind to and activate the HSP90β promoter [85][76]. In Figure 1 thwe researchers hhave summarized the crosstalking between immune cells and bone tissue in multiple myeloma.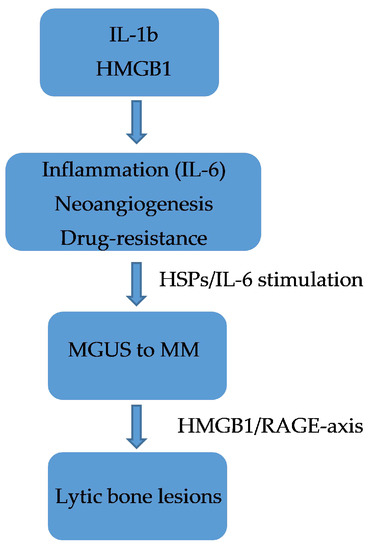
Figure 1.
Crosstalking between immune cells and bone tissue in multiple myeloma.